
Abstract
Heat shock protein 90 (Hsp90) facilitates maturation and stability of HER2. Combining an Hsp90 inhibitor and trastuzumab has demonstrated anti-tumor effects in patients with HER2+ breast cancer. Adults with measurable, locally advanced or metastatic HER2+ breast cancer and prior trastuzumab treatment were enrolled in a phase 2 trial employing weekly 300 mg/m2 retaspimycin HCl, a potent Hsp90 inhibitor, with 6 mg/kg trastuzumab every 3 weeks. A Simon’s two-stage design determined trial expansion by dose-limiting toxicity (DLT) and response rates. Pharmacokinetics and electrocardiograms were evaluated. Twenty-six patients with median age 52.5 years (range 33–72) enrolled with a median of six prior chemotherapeutic regimens (range 2–20). On study, patients received a median of three treatment cycles (range 1–12). No DLTs were observed. Most adverse events (AEs) were grade 1 or 2; common treatment-related AEs included fatigue (46 %), nausea (31 %), and diarrhea (23 %). One patient had treatment-related serious AEs of grade 1 diarrhea and grade 3 hypokalemia. grade 3 transaminase elevation occurred in one patient (4 %) who also had metastatic liver disease. Sixteen patients (62 %) had stable disease, with a median on-study duration of 2.4 months (range 1.1–8.2). No confirmed responses were observed. Retaspimycin HCl at 300 mg/m² weekly in combination with trastuzumab was well tolerated and without significant toxicities. Modest clinical activity was observed, but did not meet criteria for trial expansion. The safety profile for patients on study raises the possibility of retaspimycin HCl underdosing that limited efficacy. Studies employing higher doses are ongoing.
Similar content being viewed by others


Introduction
Preclinical models and clinical studies in patients with advanced HER2+ disease have shown that trastuzumab-refractory HER2+ breast cancer is susceptible to the effects of Heat shock protein 90 (Hsp90) inhibition [1, 2]. This effect is mediated by Hsp90 inhibition-induced HER2 degradation and, additionally, via effects on component proteins of the PI3 k-Akt pathway, p95, and HER3, all of which are implicated in the development of trastuzumab resistance [3, 4].
The most promising activity to date has been observed with tanespimycin (17AAG; KOS953), a geldanamycin analog, when given in combination with trastuzumab to HER2+ patients pre-treated and progressing on trastuzumab therapy [2, 5]. In spite of this activity, the clinical development of tanespimycin was suspended in 2010 by the sponsor and all trials with this drug have closed.
Retaspimycin hydrochloride (HCl) is an Hsp90 inhibitor designed to preserve the structural complexity of the natural parent compound while overcoming the limitations of earlier ansamycins. Because of the better aqueous solubility of retaspimycin HCl, it emerged as a potential replacement for tanespimycin. In the circulation, retaspimycin HCl is deprotonated and the free base hydroquinone is oxidized to 17-AAG; 17-AAG is subsequently reduced back to the hydroquinone by cellular reductase enzymes, such that the two moieties exist in a dynamic equilibrium in vivo.
In pre-clinical studies, retaspimycin HCl has shown promising activity in multiple models of solid and hematologic malignancies [6–9]. Specifically, retaspimycin HCl markedly reduced total levels of HER2 and Akt, as well as phosphorylated Akt and MAPK to an equal extent in trastuzumab-sensitive and trastuzumab-resistant cells. When administered either as a single agent or in combination with trastuzumab, retaspimycin HCl also inhibited in vivo the growth of both trastuzumab-sensitive and trastuzumab-resistant tumor xenografts.
Based on these pre-clinical results, and using doses identified in phase 1 clinical trials of retaspimycin HCl monotherapy, a phase 2 clinical trial was initiated to evaluate the efficacy of retaspimycin HCl plus trastuzumab in patients with advanced HER2+ metastatic breast cancer progressing on prior trastuzumab therapy.
Patients and methods
Patients
Adult patients (≥18 years of age) with pathologically confirmed locally advanced or metastatic breast cancer and one or more measurable lesions according to response evaluation criteria in solid tumors (RECIST) were enrolled. All patients were required to have HER2− expressing primary or metastatic tumors defined as either a 3+ score by immunohistochemistry or amplification on a fluorescence in situ hybridization assay. Patients must have received at least two prior regimens in the advanced setting containing a HER2− targeted agent. Trastuzumab must have been a component of at least one of these regimens. There was no upper limit to the number of prior therapies patients may have received, although patients must have had disease progression during or since their most recent systemic therapy. Additional eligibility criteria included a left ventricular ejection fraction within the normal limits and adequate baseline organ and marrow function, specifically a bilirubin within normal limits and alanine aminotransferase and aspartate aminotransferase ≤1.5× upper limit of normal (ULN).
Key exclusion criteria included prior treatment with any Hsp90 inhibitor, prior significant intolerability or adverse events (AEs) related to trastuzumab (including infusion reactions or heart failure), baseline QTc >470 ms, sinus bradycardia, or other significant co-morbidity within the previous 6 months that may have put the patient at undue risk during the study. Patients were also ineligible if they had a recent history (within 3 months) of active keratitis or keratoconjunctivitis, clinically active brain metastases, prior hepatic resections or hepatic-directed therapies, or had used medication or food that was a clinically relevant CYP3A inhibitor or inducer within 2 weeks prior to the first dose of study treatment.
All patients provided written informed consent. The study was approved by local institutional review boards and conducted in accordance with good clinical practice guidelines and the declaration of Helsinki.
Study design and assessments
This prospective, multicenter, phase 2 trial commenced in 2009. Patients were scheduled to receive retaspimycin HCl at a dose of 300 mg/m2 once or twice weekly plus trastuzumab. Patients on the twice weekly schedule received retaspimycin HCl for 2 weeks followed by 1 week off treatment. Patients on the once weekly schedule did not have an off-treatment period. After three patients enrolled on the twice weekly regimen, this schedule was closed to accrual and all new patients were enrolled at a dose of 300 mg/m2 once weekly, based upon findings in other concurrent studies with retaspimycin HCl wherein imbalanced mortality, regardless of causality, compared to placebo was observed at doses given twice weekly. Discussion of safety and efficacy is limited to the 26 patients who received treatment on the once weekly regimen, because this larger study population generated more meaningful results than the study population that received twice weekly dosing (n = 3).
Evaluable patients were defined as patients who either completed Cycle 1 or had a dose-limiting toxicity in Cycle 1, where completion of Cycle 1 was defined for the once weekly schedule as receiving one dose of trastuzumab and at least two doses of retaspimycin HCl. For the twice weekly schedule, completion of Cycle 1 was defined as receiving one dose of trastuzumab and at least three doses of retaspimycin HCl. The study was designed to pause between stages to complete safety and efficacy analyses. All patients received conventional trastuzumab every 3 weeks as an intravenous infusion of 6 mg/kg, with an 8 mg/kg initial loading dose for patients whose last trastuzumab therapy was >4 weeks prior to study entry. On days where both drugs were given together, trastuzumab was administered first.
The primary endpoints were the overall response rate (ORR), as defined by RECIST 1.1 and the evaluation safety and tolerability. Secondary endpoints included progression-free survival (PFS), time to treatment progression (TTP), overall survival (OS) and pharmacokinetics (PK). Exploratory endpoints were time to first response, duration of response, and clinical benefit consisting of a composite of complete response, partial response, tumor regression of 20–29 %, and stable disease ≥ 6 months).
Assessments and data collection
Tumor assessments and echo/multi-gated acquisition scans were made at baseline and after every two cycles (6 weeks). An independent radiographic tumor assessment with two reviewers and one adjudicator was performed in addition to local assessments. Electrocardiograms were taken at baseline and in triplicate pre- and post-infusion on day 1 of Cycles 1, 3, and 5. The NCI common terminology criteria for adverse events version 3.0 was used to grade toxicities during the trial. Given the hepatotoxicity reported in a previous study in patients with gastrointestinal stromal tumors (GIST), liver function tests (AST, ALT, total bilirubin, and alkaline phosphatase) were drawn and reviewed within 24 h prior to each dose of retaspimycin HCl. Delays and/or modifications in dose were implemented for increases in liver function test levels ≥grade 2. An ophthalmologic exam was conducted at baseline for all patients and, as needed, while patients were on study.
Dose-limiting toxicities were defined as any of the following events occurring during the first cycle of treatment that were attributable to the study regimen: death related to investigational regimen; grade 4 neutropenia lasting ≥5 days; grade ≥ 3 febrile neutropenia with elevated temperature (≥101.5 °F) on two occasions; grade 4 thrombocytopenia or anemia; grade ≥ 3 AST, ALT, alkaline phosphatase or total bilirubin at any time; grade 2 AST, ALT, or total bilirubin occurring within 24 h prior to retaspimycin HCl infusion; grade ≥ 3 diarrhea, nausea, or vomiting despite optimal medical therapy; grade ≥ 3 QTc prolongation; and, any non-hematological toxicity of ≥grade 3, except for asymptomatic lipase increases in the absence of clinical signs and symptoms of pancreatitis.
Blood samples for the main PK analyses were collected pre- and post-trastuzumab and pre- and post-retaspimycin HCl, as well as 30 min, 1, 2, 3, 5, and 24 h after dose 1 retaspimycin HCl during Cycle 1 and dose 4 of Cycle 1 for the twice weekly schedule. Additional samples at a variety of pre-specified time points during Stage 1 were also collected.
Statistical assessments
A Simon’s 2-stage optimal design was implemented within each treatment schedule with the assumed response rate for the null hypothesis of po = 11 %, the assumed response rate for the alternative hypothesis of p1 = 24 %, a targeted one-sided alpha = 0.05, and power = 80 %. If <2/20 patients achieved a response (PR or CR per RECIST 1.1) using a central independent radiology review in Stage 1, then the treatment would be deemed futile and no additional patients would be enrolled into Stage 2 for that treatment schedule. For either schedule, if criteria were met to proceed to Stage 2, then ~26 additional patients would be added to that treatment schedule and if 9 or more of the total 46 patients achieved a response, then the study would be considered successful.
Efficacy and safety analyses were conducted on the 26 patients who received treatment on the once weekly regimen, because analysis of this larger study population (n = 26) generated more statistically meaningful results than the study population that received twice weekly dosing (n = 3). Pharmacokinetic analyses were conducted on all patients with available data in the once weekly treatment regimen (n = 24) and twice weekly treatment regimen (n = 3). Patients who discontinued the study without any post-baseline disease evaluation were considered treatment failures. All efficacy analyses were to be performed for each treatment schedule separately.
Progression-free survival (PFS) defined as time to death or progression, TTP, and OS were calculated from start of treatment. Pharmacokinetic parameters based on the actual sample collection times were determined using standard noncompartmental methods. The PK parameters assessed included: Cmax, Tmax, AUC∞, T1/2, CL, and Vss. Plasma retaspimycin, 17-AAG, 17-AG, and trastuzumab concentration data and all derived PK parameters were summarized using descriptive statistics including N, mean, median, standard deviation, geometric mean, coefficient of variance, and minimum and maximum.
Results
Patients
Initially, three patients were enrolled to receive 300 mg/m2 retaspimycin HCl on the twice weekly regimen. Based on toxicity findings from another ongoing trial of retaspimycin HCl, this schedule was closed to further accrual and all subsequently enrolled patients received 300 mg/m2 retaspimycin HCl on the once weekly regimen (see study design and assessments). Results are presented for the 26 patients (25 females and 1 male) who received retaspimycin HCl on the once weekly regimen.
Overall, study patients (N = 26) were heavily pre-treated prior to entering the study. Median duration of trastuzumab therapy was 33 months (range 10–104 months), and patients had been exposed to a median of 6 prior chemotherapy regimens for breast cancer (range 2–26 regimens). Other baseline characteristics are presented in Table 1. All patients discontinued from the study for either radiographic or clinical disease progression (n = 22, 85 %) or an adverse event (n = 3, 12 %), with only one patient discontinuing for an unspecified reason.
Study treatment
Among patients who received 300 mg/m2 retaspimycin HCl once weekly (N = 26), the median number of treatment cycles initiated was three (range, 2–12 cycles) and the median number of doses of retaspimycin HCl was 7.5 (range, 4–35 doses). The median number of doses of trastuzumab (6 mg/kg every 3 weeks) was three (range, 2–11 doses). Overall, the median number of days on study therapy was 48 (range, 23–246 days).
Efficacy
In this heavily pre-treated population, the best response observed was stable disease in 16 patients (62 %), including one patient with prolonged (>6 months) stable disease (Figs. 1, 2). Patients who had stable disease received a median of ~4 cycles of treatment (range, 2–12 cycles), with a median of ~2.4 months on study (range, 1.1–8.2 months) (Fig. 2). Of the 26 patients enrolled to receive weekly dosing, 22 patients had measurable disease according to central review. Tumor shrinkage and best response in this group as determined by an independent radiographic assessment with 2 reviewers and one adjudicator (as needed) is provided in Fig. 1.

Best %-change in target lesion size. 22 patients had measurable disease according to central review, 15 (68 %) with best response of stable disease. Two patients with decreases in target lesion size <20 % had progression of disease in non-target lesions

Time on study. Patients who had stable disease received a median of ~4 cycles of treatment (range, 2–12 cycles), with a median of ~2.4 months on study (range, 1.1–8.2 months)
Safety and tolerability
The adverse events occurring in ≥5 % of patients attributable to the study treatment (retaspimycin HCL, trastuzumab, or both) are summarized in Table 2. Overall, the most common toxicities were fatigue (46 %), nausea (31 %), discolored urine (23 %), and diarrhea (23 %), mostly with CTCAE severity grades of 1 or 2. Two fatalities that were attributed to underlying disease and unrelated to either study treatment involved events of pneumonia and respiratory failure in one patient and bronchoaspiration in the second patient. Serious, related treatment-emergent AEs of grade 1 diarrhea and resulting grade 3 hypokalemia occurred in one patient. The events resolved with medical therapy and did not recur on rechallenge at 225 mg/m2 once weekly. Three patients required dose reductions due to gastrointestinal toxicity. Three additional patients discontinued therapy for treatment-related AEs: grade 2 decrease in left ventricular ejection fraction (n = 1), grade 3 increase in serum lipase (n = 1), and grade 3 increases in ALT, AST, and gamma-glutamyl transpeptidase in 1 patient with liver metastases, whose ALT and AST levels recovered to grade 2 by 30 days after discontinuing study treatment. For the majority of patients, liver enzymes remained at levels observed at baseline, whether normal or elevated. There were no cases of symptomatic ocular toxicity.
Electrocardiogram
The median baseline QTc interval using fridericia’s correction formula (QTcF) was 401.7 ms (range, 351–464). The median first post-baseline QTcF was 402.0 ms (range, 364–475), representing a median change from baseline of 3.8 ms (range, 15–34); the last “on treatment” median QTcF was 417.8 ms (range, 368–475), representing a median change of 7.5 ms (range, −14–55). The minimum and maximum values for any patient at any post-treatment time point were 364 and 475 ms, respectively. Seven (27 %) patients with normal QTcF durations at baseline had post-baseline QTcF durations >450 ms or that increased >30 ms from baseline. Overall, the QTcF values fluctuated within a narrow range, remaining ≤grade 2 at all times.
Pharmacokinetics
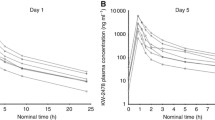
Plasma samples were analyzed for retaspimycin and its metabolites, 17-AAG, and 17-AG, and were available from 24 patients who received retaspimycin HCI once weekly and from 3 patients on the twice weekly regimen. Plasma concentrations for retaspimycin, 17-AAG, and 17-AG following administration of retaspimycin HCI once or twice weekly were similar. On Cycle 1 dose 1, the overall mean exposures (AUC0-∞) to retaspimycin, 17-AAG, and 17-AG were 10,003, 15,334, and 22,975 ng h/mL, respectively. On Cycle 4 dose 1, AUC0-∞ values for retaspimycin, 17-AAG, and 17-AG were 6,714, 14,337, and 17,261 ng h/mL, respectively. There was significant inter-patient variability in Cmax and AUC0-∞ across all analytes evaluated, with coefficient of variation (%CV) values ranging from 39 to 103 %. Exposure to 17-AG (which has similar potency to retaspimycin HCl in cellular assays) accounted for ~50 % of the total exposure to drug, calculated as the aggregate AUC of parent drug plus the two metabolites. The mean apparent terminal elimination half-lives for retaspimycin, 17-AAG, and 17-AG were 2.86, 4.86, and 4.57 h after single dose administration, and 3.40, 4.33, and 4.63 h following administration of repeated doses. Systemic clearance of retaspimycin averaged 106.5 and 94.9 L/h, on Cycle 1 day 1 and Cycle 1 day 4, respectively; however, notable variability was observed (%CV of 133 and 72). The clearance and half-life estimates obtained in this study are similar to data presented previously [10] for retaspimycin HCI. Overall, the PK parameters after repeat doses of retaspimycin HCI in the presence of trastuzumab on Cycle 4 dose 1 were similar to those observed after the first dose without trastuzumab on Cycle 1 dose 1.
Blood samples were collected for determination of trastuzumab pharmacokinetics in Cycle 4 (N = 11). Mean trastuzumab serum concentrations peaked at the end of the 90 min infusion and remained fairly flat through the first 24 h following infusion. Concentrations declined slowly thereafter, with detectable concentrations (lower limit of the bioanalytical assay = 10 μg/mL) in 6 of 6 patients who had samples collected 2 weeks after the infusion and in 4 of 5 patients who had samples collected 3 weeks after infusion. In Cycle 4, the mean trastuzumab exposure over the 24 h following dose administration was 3,094 μg*h/mL, which increased to a mean concentration of 17,322 μg*h/mL over the 3-week sampling interval. Mean systemic clearance and volume of distribution were low, estimated as 20.2 ± 7.2 mL/h and 3.9 ± 0.59 L, respectively. Based on the sampling scheme employed in this study, the mean apparent terminal elimination half-life was 154 h. It has been previously reported that sampling within a single dosing interval underestimates the terminal half live of trastuzumab [11]. This is supported by a terminal half-life estimate of 28.5 days obtained using population PK methods [12]. Direct comparison of trastuzumab PK profiles across clinical studies is challenging due to disparate patient populations, varying sample collection schedules and different PK analysis methods; however, the pharmacokinetics of trastuzumab in this combination regimen are within the range of data described in the literature [11, 13, 14].
Discussion
In previous breast cancer models in the treatment of HER2+, the combination of an Hsp90 inhibitor plus trastuzumab showed synergistic anticancer activity; this translated clinically to an ORR of 22 % and CBR of 59 % for tanespimycin (17AAG) plus trastuzumab in patients with trastuzumab-refractory disease. Despite the promise of these earlier results, the current trial in patients with HER2+ breast cancer did not meet the pre-specified expansion criteria for the combination of retaspimycin HCl plus trastuzumab. In view of PK data that show retaspimycin HCl is capable of interconversion to 17AAG in vivo, the findings from this trial are unexpected and raise important questions regarding the development of this inhibitor.
An important finding from this trial is an apparent lack of moderate or severe toxicity. While the observed favorable toxicity profile of retaspimycin HCl in this trial was supported by low rates of expected drug-related AEs, in this case, the toxicity rates might suggest that an insufficient dose of study drug was administered to adequately treat disease. The original planned dose of retaspimycin HCl for the study was 300 mg/m2 twice weekly for 2 of 3 weeks, which was lower than the twice weekly dose used in the phase 1 study (400 mg/m2) in patients with GIST. In that phase 1 study, investigators reported metabolic responses in patients with refractory GIST. This same schedule was then chosen for the follow-up phase 3 randomized GIST trial; however, an imbalance in deaths in the retaspimycin HCl arm compared to the placebo arm resulted in a decision to modify the retaspimycin HCl dose for the present trial in breast cancer to 300 mg/m2 weekly. By comparison, the recommended phase 2 dose for tanespimycin in combination with trastuzumab for the same patient population was 450 mg/m2 weekly, a dose that was also below the MTD, but that was felt to be clinically optimal because it produced tumor responses and was well tolerated. While the decision to decrease the dose in the present breast cancer trial was a necessary and appropriate action to preclude potential toxicity, the magnitude of the reduction for a breast cancer population may have been too large. Unlike the GIST population, where liver metastases can be more extensive and are often treated with site-specific therapies, including surgical liver resections and hepatic arterial chemo-embolization, patients with metastatic breast cancer are largely treated with systemic therapies and, as such, have a greater baseline liver reserve. Indeed, only one patient on the present breast cancer trial developed greater than a grade 1 liver enzyme elevation. Similarly, a comparison of other toxicities between the present breast cancer trial and that of the tanespimycin + trastuzumab trial, reveals an almost twofold increase in the incidence of toxicities seen with tanespimycin compared to retaspimycin HCl, the vast majority of which were grades 1 and 2. Overall, the absence of toxicities combined with a large number of patients with short-lived stable disease, suggests that the combination of retaspimycin HCl plus trastuzumab is active but, perhaps, was not optimally delivered in this trial.
Other ongoing trials with retaspimycin HCl are examining novel combinations in patients with advanced non-small cell lung cancer (NSCLC), where mutated EGFR, Kras and the EML4-ALK fusion protein are known sensitive clients of HSP90. In a phase 1b trial, retaspimycin HCl and docetaxel administered both weekly and on a 3 week schedule did not reveal an MTD. An expansion phase of this trial has tested retaspimycin HCl 300 mg/m2 with docetaxel given weekly with positive safety results. Results from this study led to an ongoing phase 2 evaluation of 450 mg/m2 retaspimycin HCl administered weekly in combination with 75 mg/m2 docetaxel administered every 3 weeks in patients with advance NSCLC. In addition, a phase 1 dose escalation study of retaspimycin HCl with everolimus is underway in patients with advanced NSCLC.
Second generation non-ansamycin Hsp90 inhibitors including NVP- AUY922, STA-9090/ganetespib, ATI-13387, and PU-H71, among others, are also undergoing clinical evaluation. Initial results in breast cancer, as well as other cancer types, reveal evidence of antitumor effects and the potential for an improved therapeutic index. Hence, further studies of these agents as monotherapy and in novel combinations are also planned.
Based on the results from this study, a trial in patients with HER2+ breast cancer utilizing a higher dose of retaspimycin HCl is warranted. Further, the advantages in formulation, and hence a therapeutic index with this Hsp90 inhibitor that maximizes activity and minimizes toxicity, were insufficiently tested in the current study. As is evident from prior experience and emerging patterns of drug development, any future trial must incorporate tumor biopsies or novel methods to evaluate pharmacodynamic endpoints to guide patient and dose selection with Hsp90 inhibitor therapy. Earlier Hsp90 inhibitors were not able to achieve an acceptable therapeutic index as their clinical development was hindered by toxicity and formulation issues. While the initial reports with the inhibitors currently in development are encouraging, the optimal use of these agents will depend upon a full understanding of the complex role of Hsp90 in both normal and cancer cell physiology.
References
Basso AD, Solit DB, Munster PN, Rosen N (2002) Ansamycin antibiotics inhibit Akt activation and cyclin D expression in breast cancer cells that overexpress HER2. Oncogene 21:1159–1166
Modi S, Stopeck A, Linden H, Solit D, Chandarlapaty S, Rosen N et al (2011) HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res 17(15):5132–5139 Epub 2011 May 10
Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sánchez V, Chakrabarty A et al (2011) Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci USA 108(12):5021–5026 Epub 2011 Mar 8
Gerbin CS, Landgraf R (2010) Geldanamycin selectively targets the nascent form of ERBB3 for degradation. Cell Stress Chaperones 15(5):529–544 Epub 2010 Jan 19
Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, Bagatell R et al (2007) Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J Clin Oncol 25(34):5410–5417
Peng C, Li D, Li S (2007) Heat shock protein 90: a potential therapeutic target in leukemic progenitor and stem cells harboring mutant BCR-ABL resistant to kinase inhibitors. Cell Cycle 6(18):2227–2231 Epub 2007 Jul 10
Abramson JS, Chen W, Juszczynski P, Takahashi H, Neuberg D, Kutok JL et al (2009) The heat shock protein 90 inhibitor IPI-504 induces apoptosis of AKT-dependent diffuse large B-cell lymphomas. Br J Haematol 144(3):358–366 Epub 2008 Nov 13
Song D, Chaerkady R, Tan AC, García-García E, Nalli A, Suárez-Gauthier A et al (2008) Antitumor activity and molecular effects of the novel heat shock protein 90 inhibitor, IPI-504, in pancreatic cancer. Mol Cancer Ther 7(10):3275–3284
Leow CC, Chesebrough J, Coffman KT, Fazenbaker CA, Gooya J, Weng D et al (2009) Antitumor efficacy of IPI-504, a selective heat shock protein 90 inhibitor against human epidermal growth factor receptor 2-positive human xenograft models as a single agent and in combination with trastuzumab or lapatinib. Mol Cancer Ther 8(8):2131–2141 Epub 2009 Aug 11
Siegel D, Jagannath S, Vesole DH, Borello I, Mazumder A, Mitsiades C et al (2011) A phase 1 study of IPI-504 (retaspimycin hydrochloride) in patients with relapsed or relapsed and refractory multiple myeloma. Leuk Lymphoma 52(12):2308–2315 Epub 2011 Aug 18
Baselga J, Carbonell X, Castañeda-Soto NJ, Clemens M, Green M, Harvey V et al (2005) Phase II study of efficacy, safety, and pharmacokinetics of trastuzumab monotherapy administered on a 3-weekly schedule. J Clin Oncol 23(10):2162–2171
Bruno R, Washington CB, Lu JF, Lieberman G, Banken L, Klein P (2005) Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother Pharmacol 56(4):361–369 Epub 2005 May 3
Leyland-Jones B (2001) Dose scheduling–Herceptin. Oncology 61(Suppl 2):31–36 Review
Leyland-Jones B, Gelmon K, Ayoub JP, Arnold A, Verma S, Dias R et al (2003) Pharmacokinetics, safety, and efficacy of trastuzumab administered every 3 weeks in combination with paclitaxel. J Clin Oncol 21(21):3965–3971 Epub 2003 Sep
Conflicts of interest
The authors have no financial conflicts of interest to disclose. Both Eduardo Rodenas and Jill Goddard are no longer employed by Infinity Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Additional information
Research support
Support for this trial was provided by Infinity Pharmaceuticals, Inc., Cambridge, MA, USA.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited. The exclusive right to any commercial use of the article is with Springer.
About this article
Cite this article
Modi, S., Saura, C., Henderson, C. et al. A multicenter trial evaluating retaspimycin HCL (IPI-504) plus trastuzumab in patients with advanced or metastatic HER2-positive breast cancer. Breast Cancer Res Treat 139, 107–113 (2013). https://doi.org/10.1007/s10549-013-2510-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-013-2510-5