
Abstract
Objective
To assess clinical features and general health status of adult patients with mucopolysaccharidosis (MPS) VI.
Methods
This report includes the clinical history of patients older than 18 years with slowly progressing MPS VI and the retrospective analysis of the outcomes of available data collected between September 2003 and October 2008 at the Center of Pediatric and Adolescent Medicine, University Medical Center, Johannes Gutenberg-University of Mainz, Germany. Variables included were urinary glycosaminoglycan (uGAG) level, mutation analysis, body height, forced vital capacity (FVC), 6-minute walk test, echocardiographic findings, the need for craniocervical decompression surgery, orthopaedic findings and ophthalmological assessments.
Results
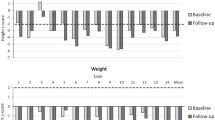
The analysis included nine patients with MPS VI aged 19-29 years. The median age at diagnosis was 12 (range 6–20) years. At the time of the assessment (median age 25 years), median uGAG was 29 (range 15-149) μg/mg creatinine and median height 152 (range 136–161) cm. All patients had a FVC below standard values, seven showed reduced endurance in the 6-minute-walk test, all had valve changes with valve replacement in three, two underwent craniocervical decompression surgery, two underwent carpal tunnel surgery, five had ear/nose/throat (ENT) interventions, seven had hip pain/dysplasia, seven had corneal clouding and two were visually impaired.
Conclusions
Although patients with slowly progressing MPS VI are a heterogeneous group showing disease manifestations in several organs, they seem to have some typical characteristics in common. Despite the attenuated clinical course, many of these patients show severe morbidity. Therefore, early diagnosis and proper follow-up and treatment are essential.

Similar content being viewed by others

Reference
Ashworth JL, Biswas S, Wraith E, Lloyd IC (2006) The ocular features of the mucopolysaccharidoses. Eye 20:553–563
Azevedo ACMM, Schwartz IV, Kalakun L et al (2004) Clinical and biochemical study of 28 patients with mucopolysaccharidosis type VI. Clin Genet 66:208–213
Baehner F, Schmiedeskamp C, Krummenauer F et al (2005) Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis 28:1011–1017
Decker C, Yu Z, Giugliani R et al (2010) Enzyme replacement therapy for mucopolysaccharidosis VI: growth and pubertal development in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Pediatr Rehabil Med 3:89–100
Enright PL, Sherrill DL (1998) Reference equations for the six-minute walk in healthy adults. Am J Respir Crit Care Med 158:1384–1387
Fesslová V, Corti P, Sersale G et al (2009) The natural course and the impact of therapies of cardiac involvement in the mucopolysaccharidoses. Cardiol Young 19:170–178
Giugliani R, Harmatz P, Wraith JE (2007) Management guidelines for mucopolysaccharidosis VI. Pediatrics 120:405–418
Gottwald I, Hughes J, Stewart F et al (2011) Attenuated mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome) due to homozygosity for the p.Y210C mutation in the ARSB gene. Mol Genet Metab 103:300–302
Harmatz P, Whitley CB, Waber L et al (2004) Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). J Pediatr 144:574–580
Harmatz P, Ketteridge D, Giugliani R et al (2005) Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4-sulfatase. Pediatrics 115:e681–e689
Harmatz P, Giugliani R, Schwartz I et al (2006) Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J Pediatr 148:533–539
Harmatz P, Yu ZF, Giugliani R et al (2010) Enzyme replacement therapy for mucopolysaccharidosis VI: evaluation of long-term pulmonary function in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Inherit Metab Dis 33:51–60
Hendriksz CJ, Giugliani R, Harmatz P et al (2011) Design, baseline characteristics and early findings of the MPS VI Clinical Surveillance Program (CSP). J Inherit Metab Dis. doi:10.1007/s10545-011-9410-9
Isbrandt D, Arlt G, Brooks DA, Hopwood JJ, von Figura K, Peters C (1994) Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): six unique arylsulfatase B gene alleles causing variable disease phenotypes. Am J Hum Genet 54:454–463
Karageorgos L, Brooks DA, Pollard A et al (2007) Mutational analysis of 105 mucopolysaccharidosis type VI patients. Hum Mutat 28:897–903
Leal GN, de Paula AC, Leone C, Kim CA (2010) Echocardiographic study of paediatric patients with mucopolysaccharidosis. Cardiol Young 20:254–261
Litjens T, Brooks DA, Peters C, Gibson GJ, Hopwood JJ (1996) Identification, expression, and biochemical characterization of N-acetylgalactosamine-4-sulfatase mutations and relationship with clinical phenotype in MPS-VI patients. Am J Hum Genet 58:1127–1134
McGill JJ, Inwood AC, Coman DJ, Lipke ML et al (2010) Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age–a sibling control study. Clin Genet 77:492–498
Mut M, Cila A, Varli K, Akalan N (2005) Multilevel myelopathy in Maroteaux-Lamy syndrome and review of the literature. Clin Neurol Neurosurg 107:230–235
Neufeld EF, Muenzer J (2001) The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 3421–3452
Scarpa M, Buffone E, La Marca P, Campello M, Rampazzo A (2010) Difficulties in diagnosing slowly progressive mucopolysaccharidosis VI: a case series. J Pediatr Rehabil Med 3:71–75
Swiedler SJ, Beck M, Bajbouj M et al (2005) Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey of subjects with mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). Am J Med Genet 134A:144–150
Thorne JA, Javadpour M, Hughes DG, Wraith E, Cowie RA (2001) Craniovertebral abnormalities in type VI mucopolysaccharidosis (Maroteaux-Lamy syndrome). Neurosurgery 48:849–853
Valayannopoulos V, Nicely H, Harmatz P, Turbeville S (2010) Mucopolysaccharidosis VI. Orphanet J Rare Dis 5:5
Wippermann CF, Beck M, Schranz D, Huth R, Michel-Behnke I, Jüngst BK (1995) Mitral and aortic regurgitation in 84 patients with mucopolysaccharidoses. Eur J Pediatr 154:98–101
Acknowledgments
The authors are grateful to Ismar Healthcare NV for their assistance in writing of the manuscript, which was funded by BioMarin Europe Ltd.
Competing interests
Dr. Elke Miebach has received travel expenses and speakers fees for scientific meetings by BioMarin Pharmaceutical Inc., Genzyme Corp. and Shire, Inc., Dr. Christina Lampe has received travel expenses and speakers fees for scientific meetings by BioMarin, Shire Inc. and Genzyme. Prof. Susanne Pitz has received travel and research grants from BioMarin, Genzyme and Shire, Inc., Dr. Wolfgang Kamin and Dr. Christoph Kampmann have received travel and research grants as well as speakers fees for scientific meetings by BioMarin, Genzyme and Shire Inc., Dr. Bianca Link has received travel and research grants as well as honoraria for scientific presentations from BioMarin, Genzyme and Shire Inc. Dr. Eugen Mengel received speakers fee and research grants from Genzyme and Shire, he acts as consultant for BioMarin respective MPS IV. Dr. Anke Thümler has no conflict of interest.
Funding
The preparation of this manuscript was supported by BioMarin Europe Ltd.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Ed Wraith
Rights and permissions
About this article
Cite this article
Thümler, A., Miebach, E., Lampe, C. et al. Clinical characteristics of adults with slowly progressing mucopolysaccharidosis VI: a case series. J Inherit Metab Dis 35, 1071–1079 (2012). https://doi.org/10.1007/s10545-012-9474-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-012-9474-1