
Abstract
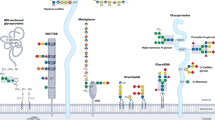
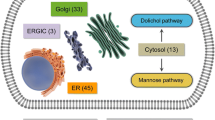
Glycoprotein biosynthesis describes the process of co- and posttranslational attachment of sugar chains to proteins, a process that has been found in nearly all known organisms. Human deficiencies evoked by mutations in the glycosylation pathway of glycoproteins lead to congenital disorders of glycosylation (CDG), a rapidly expanding group of autosomal recessive inherited metabolic diseases with multisystemic phenotypes that are mostly combined with severe neurological impairment. Although investigations on new types of CDG have proceeded rapidly in recent years, the correlation between inaccurate protein glycosylation and pathological loss of functionality of distinct organ systems remains widely unknown, and therapeutics for the patients are mostly not available. Therefore, mouse models provide an outstanding helpful tool for investigations on different aspects of glycosylation deficiencies that cannot be performed in patients or cell culture. This review focuses on existing mouse models generated for the types of CDG that affect the N-glycosylation pathway.




Similar content being viewed by others

References
Aebi M, Helenius A, Schenk B, Barone R, Fiumara A, Berger EG, Hennet T et al (1999) Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosylation: an updated nomenclature for CDG. First International Workshop on CDGS. Glycoconj J 16:669–671
Akama TO, Nakagawa H, Wong NK, Sutton-Smith M, Dell A, Morris HR, Nakayama J et al (2006) Essential and mutually compensatory roles of {alpha}-mannosidase II and {alpha}-mannosidase IIx in N-glycan processing in vivo in mice. Proc Natl Acad Sci USA 103:8983–8988
Asano M, Furukawa K, Kido M, Matsumoto S, Umesaki Y, Kochibe N, Iwakura Y (1997) Growth retardation and early death of β-1,4-galactosyltransferase knockout mice with augmented proliferation and abnormal differentiation of epithelial cells. EMBO J 16:1850–1857
Bendiak B, Schachter H (1987) Control of glycoprotein synthesis. Kinetic mechanism, substrate specificity, and inhibition characteristics of UDP-N-acetylglucosamine: alpha-D-mannoside beta 1-2 N-acetylglucosaminyltransferase II from rat liver. J Biol Chem 262:5784–5790
Cantagrel V, Lefeber DJ, Ng BG, Guan Z, Silhavy JL, Bielas SL, Lehle L et al (2010) SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell 142:203–217
Cormier-Daire V, Amiel J, Vuillaumier-Barrot S, Tan J, Durand G, Munnich A, Le Merrer M et al (2000) Congenital disorders of glycosylation IIa cause growth retardation, mental retardation, and facial dysmorphism. J Med Genet 37:875–877
D'Agostaro GA, Zingoni A, Moritz RL, Simpson RJ, Schachter H, Bendiak B (1995) Molecular cloning and expression of cDNA encoding the rat UDP-N-acetylglucosamine:alpha-6-D-mannoside beta 1, 2-N-acetylglucosaminyltransferase II. J Biol Chem 270:15211–15221
de Lonlay P, Seta N (2009) The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib. Biochim Biophys Acta 1792:841–843
DeRossi C, Bode L, Eklund EA, Zhang F, Davis JA, Westphal V, Wang L et al (2006) Ablation of mouse phosphomannose isomerase (Mpi) causes mannose 6-phosphate accumulation, toxicity, and embryonic lethality. J Biol Chem 281:5916–5927
Engelhardt H, Staudt M, Hassler A, Holzbach U, Freisinger P, Krageloh-Mann I (1999) Carbohydrate-deficient glycoprotein syndrome type 2. J Inherit Metab Dis 22:192–193
Etzioni A, Frydman M, Pollack S, Avidor I, Phillips ML, Paulson JC, Gershoni-Baruch R (1992) Brief report: recurrent severe infections caused by a novel leukocyte adhesion deficiency. N Engl J Med 327:1789–1792
Freeze HH, Sharma V (2010) Metabolic manipulation of glycosylation disorders in humans and animal models. Semin Cell Dev Biol 21:655–662
Grünewald S (2009) The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia). Biochim Biophys Acta 1792:827–834
Haeuptle MA, Hennet T (2009) Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat 30:1628–1641
Hansske B, Thiel C, Lübke T, Hasilik M, Höning S, Peters V, Heidemann PH et al (2002) Deficiency of UDP-galactose:N-acetylglucosamine beta-1,4-galactosyltransferase I causes the congenital disorder of glycosylation type IId. J Clin Invest 109:725–733
Hellbusch CC, Sperandio M, Frommhold D, Yakubenia S, Wild MK, Popovici D, Vestweber D et al (2007) Golgi GDP-fucose transporter-deficient mice mimic congenital disorder of glycosylation IIc/leukocyte adhesion deficiency II. J Biol Chem 282:10762–10772
Hidalgo A, Ma S, Peired AJ, Weiss LA, Cunningham-Rundles C, Frenette PS (2003) Insights into leukocyte adhesion deficiency type 2 from a novel mutation in the GDP-fucose transporter gene. Blood 101:1705–1712
Ioffe E, Stanley P (1994) Mice lacking N-acetylglucosaminyltransferase I activity die at mid-gestation, revealing an essential role for complex or hybrid N-linked carbohydrates. Proc Natl Acad Sci USA 91:728–732
Jaeken J, Schachter H, Carchon H, de Cock P, Coddeville B, Spik G (1994) Carbohydrate deficient glycoprotein syndrome type II: a deficiency in Golgi localised N-acetyl-glucosaminyltransferase II. Arch Dis Child 71:123–127
Jaeken J, Hennet T, Matthijs G, Freeze HH (2009) CDG nomenclature: time for a change! Biochim Biophys Acta 1792:825–826
Kumar R, Stanley P (1989) Transfection of a human gene that corrects the Lec1 glycosylation defect: evidence for transfer acetylglucosamilyltransferase I. Mol Cell Biol 9:5713–5717
Kumar R, Yang J, Larsen RD, Stanley P (1990) Cloning and expression of N-acetylglucosaminyltransferase I, the medial Golgi transferase that initiates complex N-linked carbohydrate formation. Proc Natl Acad Sci USA 87:9948–9952
Lu Q, Hasty P, Shur BD (1997) Targeted mutation in β1,4-galactosyltransferase leads to pituitary insufficiency and neonatal lethality. Dev Biol 181:257–267
Lübke T, Marquardt T, von Figura K, Körner C (1999) A new type of carbohydrate-deficient glycoprotein syndrome due to a decreased import of GDP-fucose into the golgi. J Biol Chem 274:25986–25989
Lübke T, Marquardt T, Etzioni A, Hartmann E, von Figura K, Körner C (2001) Complementation cloning identifies CDG-IIc, a new type of congenital disorders of glycosylation, as a GDP-fucose transporter deficiency. Nat Genet 28:73–76
Lühn K, Wild MK, Eckhardt M, Gerardy-Schahn R, Vestweber D (2001) The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat Genet 28:69–72
Marek KW, Vijay IK, Marth JD (1999) A recessive deletion in the GlcNAc-1-phosphotransferase gene results in peri-implantation embryonic lethality. Glycobiology 9:1263–1271
Marquardt T, Lühn K, Srikrishna G, Freeze HH, Harms E, Vestweber D (1999) Correction of leukocyte adhesion deficiency type II with oral fucose. Blood 94:3976–3985
Matthijs G, Schollen E, Pardon E, Veiga-Da-Cunha M, Jaeken J, Cassiman JJ, Van Schaftingen E (1997) Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nat Genet 16:88–92
Metzler M, Gertz A, Sarkar M, Schachter H, Schrader JW, Marth JD (1994) Complex asparagine-linked oligosaccharides are required for morphogenic events during post-implantation development. EMBO J 13:2056–2065
Morava E, Wevers RA, Cantagrel V, Hoefsloot LH, Al-Gazali L, Schoots J, van Rooij A et al (2010) A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism. Brain 133:3210–3220
Niehues R, Hasilik M, Alton G, Körner C, Schiebe-Sukumar M, Koch HG, Zimmer KP et al (1998) Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J Clin Invest 101:1414–1420
Peters V, Penzien JM, Reiter G, Körner C, Hackler R, Assmann B, Fang J et al (2002) Congenital disorder of glycosylation IId (CDG-IId)—a new entity: clinical presentation with Dandy-Walker malformation and myopathy. Neuropediatrics 33:27–32
Rabionet M, van der Spoel AC, Chuang CC, von Tümpling-Radosta B, Litjens M, Bouwmeester D, Hellbusch CC et al (2008) Male germ cells require polyenoic sphingolipids with complex glycosylation for completion of meiosis: a link to ceramide synthase-3. J Biol Chem 283:13357–13369
Sarkar M, Hull E, Nishikawa Y, Simpson RJ, Moritz RL, Dunn R, Schachter H (1991) Molecular cloning and expression of cDNA encoding the enzyme that controls conversion of high-mannose to hybrid and complex N-glycans: UDP-N-acetylglucosamine:alpha-3-D-mannoside beta-1,2-N-acetylglucosaminyltransferase I. Proc Natl Acad Sci USA 88:234–238
Schachter H (1991) The “yellow brick road'' to branched complex N-glycans. Glycobiology 1:453–461
Tan J, Dunn J, Jaeken J, Schachter H (1996) Mutations in the MGAT2 gene controlling complex N-glycan synthesis cause carbohydrate-deficient glycoprotein syndrome type II, an autosomal recessive disease with defective brain development. Am J Hum Genet 59:810–817
Thiel C, Lübke T, Matthijs G, von Figura K, Körner C (2006) Targeted disruption of the mouse phosphomannomutase 2 gene causes early embryonic lethality. Mol Cell Biol 26:5615–5620
Van Schaftingen E, Jaeken J (1995) Phosphomannomutase deficiency is a cause of carbohydrate-deficient glycoprotein syndrome type I. FEBS Lett 377:318–320
Wang Y, Tan J, Sutton-Smith M, Ditto D, Panico M, Campbell RM, Varki NM (2001) Modeling human congenital disorder of glycosylation type IIa in the mouse: conservation of asparagine-linked glycan-dependent functions in mammalian physiology and insights into disease pathogenesis. Glycobiology 11:1051–1070
Wang Y, Schachter H, Marth JD (2002) Mice with a homozygous deletion of the Mgat2 gene encoding UDP-N-acetylglucosamine:alpha-6-D-mannoside beta1,2-N-acetylglucosaminyltransferase II: a model for congenital disorder of glycosylation type IIa. Biochim Biophys Acta 1573:301–311
Wu X, Rush JS, Karaoglu D, Krasnewich D, Lubinsky MS, Waechter CJ, Gilmore R et al (2003) Deficiency of UDP-GlcNAc:dolichol phosphate N-acetylglucosamine-1 phosphate transferase (DPAGT1) causes a novel congenital disorder of glycosylation type Ij. Hum Mutat 22:144–150
Yakubenia S, Frommhold D, Schölch D, Hellbusch CC, Körner C, Petri B, Jones C et al (2008) Leukocyte trafficking in a mouse model for leukocyte adhesion deficiency II/congenital disorder of glycosylation IIc. Blood 112:1472–1481
Ye Z, Marth JD (2004) N-glycan branching requirement in neuronal and post-natal viability. Glycobiology 14:547–558
Acknowledgments
The authors of this article were supported by the Deutsche Forschungsgemeinschaft and the Fritz Thyssen Stiftung.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Jaak Jaeken
Competing interest: None declared.
Approval from the Institutional Committee for Care and Use of Laboratory Animals: the authors certify that an approval from the Institutional Committee for Care and Use of Laboratory Animals was not required.
Rights and permissions
About this article
Cite this article
Thiel, C., Körner, C. Mouse models for congenital disorders of glycosylation. J Inherit Metab Dis 34, 879–889 (2011). https://doi.org/10.1007/s10545-011-9295-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-011-9295-7