
Summary
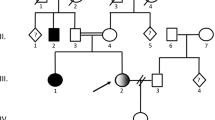
Late-onset neurological disease has rarely been reported in patients with glutaryl-CoA dehydrogenase (GCDH) deficiency. We present two siblings with GCDH deficiency. One of them presented with the classic neurological disease (patient 1). Routine investigation of family members revealed that her apparently unharmed 13-year-old sister was also affected (patient 2). Patient 2 started to have academic difficulties in the months prior to our assessment. Her clinical examination was normal, with the exception of a cranial circumference of 57 cm (slightly over the 98th centile). A severe leukoencephalopathy was demonstrated on MRI. Neuropsychological assessment showed an IQ within the normal–low range and a mild impairment of memory and executive function. Previous reports on late-onset neurological disease in GCDH deficiency have revealed that progressive leukoencephalopathy develops over time. Following the recently published guideline for the diagnosis and management of GCDH deficiency, both patients are receiving dietary treatment in combination with l-carnitine supplementation. We emphasize the need to search for chronic neurological changes of late-onset type in apparently unaffected GCDH deficiency cases diagnosed in routine family investigations.
Similar content being viewed by others


Author information
Authors and Affiliations
Corresponding author
Additional information
Communicating editor: Georg Hoffmann
Online citation: JIMD Short Report #091 (2007) Online
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
López-Laso, E., García-Villoria, J., Martín, E. et al. Classic and late-onset neurological disease in two siblings with glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 30, 979 (2007). https://doi.org/10.1007/s10545-007-0699-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-007-0699-3