
Summary
Background
A significant percentage of patients with hyperphenylalaninaemia (HPA) due to primary deficiency of the phenylalanine hydroxylase enzyme (PAH) respond to a dose of tetrahydrobiopterin (BH4) with an increased rate of phenylalanine (Phe) disposal. The effect is exploited therapeutically, with some patients on BH4 even tolerating a normal diet.
Aim
Classification of the Phe blood level response to a BH4 load by percentage reduction (PR) suffers from loss of information: only part of usually more extensive test data is used, and PR values for different times after load cannot be compared directly. Calculation of half-life (t 1/2) of blood Phe is proposed as an alternative. This classic measure unifies interpretation of tests of different duration (e.g. 8 or 15 h). t 1/2 subsumes first-order formation of tyrosine, of Phe metabolites, and renal Phe excretion; zero-order net protein synthesis can be neglected during short-time tests.
Method
t 1/2 is easily and robustly obtained by fit-ting the total set of (3–4) data points to a log-linear regression.
Results
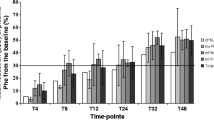
The advantage of calculating t 1/2 is exemplified by the analysis of selected published data. The results clearly speak in favour of an 8 h test period because so-called ‘slow’ responders could also be detected within this time window and because tests of longer duration are less reliable kinetically. Sequential Phe and Phe/BH4 loading tests appear advantageous because the ‘natural’ t 1/2 (without supplementation of BH4) is not normally known beforehand.
Conclusion
With t 1/2 as a reliable parameter of BH4 responsiveness, therapeutic decisions would be more rational and genotype–phenotype analysis may also profit.
Similar content being viewed by others

References
Bartholomé K (1974) Letter: A new molecular defect in phenylketonuria. Lancet 2 (7896): 1580.
Bernegger C, Blau N (2002) High frequency of tetrahydrobiopterin-responsiveness among hyperphenylalaninemias: a study of 1919 patients observed from 1988 to 2002. Mol Genet Metab 77: 304–313.
Bélanger-Quintana A, Garcia MJ, Castro M, et al (2005) Spanish BH4-responsive phenylalanine hydoxylase-deficient patients: Evolution of seven patients on long-term treatment with tetrahydrobiopterin. Mol Gen Metab 86(Supplement 1): S61–S66.
Blau N, Erlandsen H (2004) The metabolic and molecular bases of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Mol Genet Metab 82: 101–111.
Boneh A, Francis DM, Humphrey M, et al (2006) Three-year audit of the hyperphenylalaninaemia/phenylketonuria spectrum in Victoria. J Pediatr Child Health 42: 496–498.
Curtius HC, Niederwieser A, Viscontini M, et al (1979) Atypical phenylketonuria due to tetrahydrobiopterin deficiency. Diagnosis and treatment with tetrahydrobiopterin, dihydrobiopterin and sepiapterin. Clin Chim Acta 93: 251–262.
Danks DM, Bartholomé K, Clayton BE, et al (1978) Malignant hyperphenylalaninaemia – Current status (June 1977). J Inherit Metab Dis 1: 49–53.
Danks DM, Cotton RGH, Schlesinger P (1979) Diagnosis of malignant hyperphenylalaninemia. Arch Dis Child 54: 329–330.
Desviat LR, Pérez B, Bèlanger-Quintana A, et al (2004) Tetrahydrobiopterin responsiveness: results of the BH4 loading test in 31 Spanish PKU patients and correlation with their genotype. Mol Genet Metab 83: 157–162.
Erlandsen H, Pey AL, Gámez A, et al (2004) Correction of kinetic and stability defects by tetrahydrobiopterin in phenylketonuria patients with certain phenylalanine hydroxylase mutations. Proc Natl Acad Sci USA 101: 16903–16908.
EMEA: European Medicines Agency, Committee for Orphan Medicinal Products (29 May 2007) Public summary of positive opinion for orphan designation of tetrahydrobiopterin for the treatment of hyperphenylalaninemia. Doc.Ref.: EMEA/COMP/258/04 Rev. 2.
Fiege B, Blau N (2007) Assessment of tetrahydrobiopterin (BH4) responsiveness in phenylketonuria. J Pediatr 150: 627–630.
Fiege B, Bonafé L, Ballhausen D, et al (2005) Extended tetrahydrobiopterin loading test in the diagnosis of cofactor-responsive phenylketonuria: A pilot study. Mol Genet Metab 86: S91–S95.
Fiori L, Fiege B, Riva E, et al (2005) Incidence of BH4-responsiveness in phenylalanine-hydroxylase-deficient Italian patients. Mol Genet Metab 86: S67–S74.
Habich MS (2006) Pharmakologische Therapie der Phenylketonurie durch Defekt der Phenylalaninhydroxylase mit Tetrahydrobiopterin: Effekt auf Metabolite, in vivo Enzymaktivität und Proteintoleranz. Med. Thesis, Ludwig-Maximilian-University, Munich.
Kure S, Hou DC, Ohura T, et al (1999) Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J Pediatr 135: 375–378.
Langenbeck U, Wendel U (1997) Kinetic analysis of phenylalanine disposal in phenylalanine hydroxylase (PAH) deficiency. Int Pediatr 12: 19–22.
Langenbeck U, Zschocke J, Wendel U, Hönig V (2001) Modelling the phenylalanine blood level response during treatment of phenylketonuria. J Inherit Metab Dis 24: 805–814.
Leatherbarrow RJ (1987) ENZFITTER. A Non-linear Regression Data Analysis Program for the IBM PC (and True Compatibles). Cambridge, UK: Elsevier-BIOSOFT.
Lindner M, Steinfeld R, Burgard P, et al (2003) Tetrahydrobiopterin sensitivity in German patients with mild phenylalanine hydroxylase deficiency. Hum Mutat 21: 400 (Mutation in brief #588).
Muntau AC, Röschinger W, Habich M, et al (2002) Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N Engl J Med 347: 2122–2132.
Niederwieser A, Ponzone A, Curtius H-Ch (1985) Differential diagnosis of tetrahydrobiopterin deficiency. J Inherit Metab Dis 8(Supplement 1): 34–38.
Porta F, MussaA, Ferraris S, et al (2007) ‘Responsiveness’ and unresponsiveness to BH4 of PAH deficiency. J Inherit Metab Dis 30(Supplement 1): 16.
Rey F, Blandin-Savoja F, Rey J (1979) Kinetics of phenylalanine disappearance after intra-venous load in phenylketonuria and its variants. Pediatr Res 13: 21–25.
Ritschel WA (1982) Handbook of Basic Pharmakokinetics, 2nd edn. Hamilton, IL: Drug Intelligence Publications.
Rosenberg LE (1976) Vitamin-responsive inherited metabolic disorders. Adv Hum Genet 6: 1–74.
Schadewaldt P, Dalle-Feste C, Langenbeck U, Wendel U (1991) Oral l-alloisoleucine loading studies in healthy subjects and in patients with maple syrup urine disease. Pediatr Res 30: 430–434.
Shintaku H, Kure S, Ohura T, et al (2004) Long-term treatment and diagnosis of tetrahydrobiopterin-responsive hyperphenylalaninemia with a mutant phenylalanine hydroxylase gene. Pediatr Res 55: 425–430.
Smith I, Lloyd J (1974) Proceedings: Atypical phenylketonuria accompanied by severe progressive neurological illness unresponsive to dietary treatment. Arch Dis Child 49: 245.
Snyderman SE, Norton PM, Roitman E, Holt LE Jr (1964) Maple syrup urine disease, with particular reference to dietotherapy. Pediatrics 34: 454–472.
Steinfeld R, Kohlschütter A, Zschocke J, Lindner M, Ullrich K, Lukacs Z (2002) Tetrahydrobiopterin monotherapy for phenylketonuria patients with common mild mutations. Eur J Pediatr 161: 403–405.
Trefz FK, Aulehla-Scholz C, Blau N (2001) Successful treatment of phenylketonuria with tetrahydrobiopterin. Eur J Pediatr 160: 315.
Trefz F, Burton B, Longo N, et al (2007) The effect of sapropterin dihydrochloride (tetrahydrobiopterin or 6R-BH4) treatment on phenylalanine (Phe) tolerance in children with phenylketonuria controlled on a Phe-restricted diet. J Inherit Metab Dis 30(Supplement 1): 17.
Zurflüh MR, Fiori L, Fiege B, et al (2006) Pharmacokinetics of orally administered tetrahydrobiopterin in patients with phenylalanine hydroxylase deficiency. J Inherit Metab Dis 29: 725–731.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicating editor: Johannes Zschocke
Competing interests: None declared
References to electronic databases: Phenylketonuria: OMIM +261600. Phenylalanine hydroxylase: EC 1.14.16.1.
Rights and permissions
About this article
Cite this article
Langenbeck, U. Classifying tetrahydrobiopterin responsiveness in the hyperphenylalaninaemias. J Inherit Metab Dis 31, 67–72 (2008). https://doi.org/10.1007/s10545-007-0572-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-007-0572-4