
Summary
Carnitine transporter deficiency (CTD) and holocarboxylase synthetase deficiency (HLCSD) are frequent in The Faroe Islands compared to other areas, and treatment is available for both disorders. In order to evaluate the feasibility of neonatal screening in The Faroe Islands we studied detection in the neonatal period by tandem mass spectrometry, carrier frequencies, clinical manifestations, and effect of treatment of CTD and HLCSD. We found 11 patients with CTD from five families and 8 patients with HLCSD from five families. The natural history of both disorders varied extensively among patients, ranging from patients who presumably had died from their disease to asymptomatic individuals. All symptomatic patients responded favourably to supplementation with l-carnitine (in case of CTD) or biotin (in case of HLCSD), but only if treated early. Estimates of carrier frequency of about 1:20 for both disorders indicate that some enzyme-deficient individuals remain undiagnosed. Prospective and retrospective tandem mass spectrometry (MS/MS) analyses of carnitines from neonatally obtained filter-paper dried blood-spot samples (DBSS) uncovered 8 of 10 individuals with CTD when using both C0 and C2 as markers (current algorithm) and 10 of 10 when using only C0 as marker. MS/MS analysis uncovered 5 of 6 patient with HLCSD. This is the first study to report successful neonatal MS/MS analysis for the diagnosis of HLCSD. We conclude that CTD and HLCSD are relatively frequent in The Faroe Islands and are associated with variable clinical manifestations, and that diagnosis by neonatal screening followed by early therapy will secure a good outcome.
Similar content being viewed by others

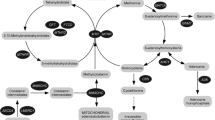
Abbreviations
- CTD:
-
carnitine transporter deficiency
- DBSS:
-
filter-paper dried blood-spot samples
- HLCS:
-
holocarboxylase synthetase
- HLCSD:
-
holocarboxylase synthetase deficiency
- MS/MS:
-
tandem mass spectrometry
References
Christensen E, Jacobsen BB, Gregersen N, et al (1981) Urinary excretion of succinylacetone and delta-aminolevulinic acid in patients with hereditary tyrosinemia. Clin Chim Acta 116: 331–341.
Christensen E, Vikre-Jorgensen J (1995) Six years’ experience with carnitine supplementation in a patient with an inherited defective carnitine transport system. J Inherit Metab Dis 18: 233–236.
Christensen E, et al (2000) Sudden infant death following pivampicillin treatment in a patient with carnitine deficiency [Abstract]. J Inherit Metab Dis 23(Supplement 1): 117.
Evans AM, Fornasini G (2003) Pharmacokinetics of l-carnitine. Clin Pharmacokinet 42: 941–967.
Lamhonwah AM, Olpin SE, Pollitt RJ, et al (2002) Novel OCTN2 mutations: no genotype–phenotype correlations: early carnitine therapy prevents cardiomyopathy. Am J Med Genet 111: 271–284.
Morrone A, Malvagia S, Donati MA, et al (2002) Clinical findings and biochemical and molecular analysis of four patients with holocarboxylase synthetase deficiency. Am J Med Genet 111: 10–18.
Narisawa K, Arai N, Igarashi Y, et al (1982) Clinical and biochemical findings on a child with multiple biotin-responsive carboxylase deficiencies. J Inherit Metab Dis 5: 67–68.
Nezu J, Tamai I, Oku A, et al (1999) Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet 21: 91–94.
Pierpont M, Breningstall GN, Stanley CA, Singh A (2000) Familial carnitine transporter defect: a treatable cause of cardiomyopathy in children. Am Heart J 139: 96–106.
Sakamoto O, Suzuki Y, Li X, et al (2000) Diagnosis and molecular analysis of an atypical case of holocarboxylase synthetase deficiency. Eur J Pediatr 159: 18–22.
Santer R, Kinner M, Steuerwald U, et al (2001) Molecular genetic basis and prevalence of glycogen storage disease type IIIA in the Faroe Islands. Eur J Hum Genet 9: 388–391.
Stanley CA, DeLeeuw S, Coates PM, et al (1991) Chronic cardiomyopathy and weakness or acute coma in children with a defect in carnitine uptake. Ann Neurol 30: 709–716.
Suzuki Y, Aoki Y, Ishida Y, et al (1994) Isolation and characterization of mutations in the human holocarboxylase synthetase cDNA. Nat Genet 8: 122–128.
Suzuki Y, Yang X, Aoki Y, Kure S, Matsubara Y (2005) Mutations in the holocarboxylase synthetase gene HLCS. Hum Mutat 26: 285–290.
Vijay S, Patterson A, Olpin S, et al (2006) Carnitine transporter defect: diagnosis in asymptomatic adult women following analysis of acylcarnitines in their newborn infants. J Inherit Metab Dis 29: 627–630.
Wang Y, Ye J, Ganapathy V, Longo N (1999) Mutations in the organic cation/carnitine transporter OCTN2 in primary carnitine deficiency. Proc Natl Acad Sci USA 96: 2356–2360.
Wilcken B, Wiley V, Sim KG, Carpenter K (2001) Carnitine transporter defect diagnosed by newborn screening with electrospray tandem mass spectrometry. J Pediatr 138: 581–584.
Yang X, Aoki Y, Li X, et al (2001) Structure of human holocarboxylase synthetase gene and mutation spectrum of holocarboxylase synthetase deficiency. Hum Genet 109: 526–534.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicating editor: Bridget Wilcken
Competing interests: None declared
References to electronic databases: CTD, OMIM 212140; SLC22A5, OMIM 603377; GenBank AC004628; HLCSD, OMIM 253270; HLCS, OMIM 609018; GenBank BC060787; EC 6.3.4.10
Rights and permissions
About this article
Cite this article
Lund, A.M., Joensen, F., Hougaard, D.M. et al. Carnitine transporter and holocarboxylase synthetase deficiencies in The Faroe Islands. J Inherit Metab Dis 30, 341–349 (2007). https://doi.org/10.1007/s10545-007-0527-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-007-0527-9