
Summary
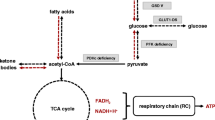
Beginning with phenylketonuria, dietary therapy for inborn errors has focused primarily on the restriction of the precursor to an affected catabolic pathway in an attempt to limit the production of potential toxins. Anaplerotic therapy is based on the concept that there may exist an energy deficit in these diseases that might be improved by providing alternative substrate for both the citric acid cycle (CAC) and the electron transport chain for enhanced ATP production. This article focuses on this basic problem, as it may relate to most catabolic disorders, and provides our current experience involving inherited diseases of mitochondrial fat oxidation, glycogen storage, and pyruvate metabolism using the anaplerotic compound triheptanoin. The observations have led to a realization that ‘inter-organ’ signalling and ‘nutrient sensors’ such as adenylate monophosphate mediated-protein kinase (AMPK) and mTOR (mammalian target of rapamycin) appear to play a significant role in the intermediary metabolism of these diseases. Activated AMPK turns on catabolic pathways to augment ATP production while turning off synthetic pathways that consume ATP. Information is provided regarding the inter-organ requirements for more normal metabolic function during crisis and how anaplerotic therapy using triheptanoin, as a direct source of substrate to the CAC for energy production, appears to be a more successful approach to an improved quality of life for these patients.
Similar content being viewed by others

References
Bodamer OA, Haas D, Hermans MM, Reuser AJ, Hoffman GF (2002) l-Alanine supplementation in late infantile glycogen storage disease type II. Pediatr Neurol 27: 145–146.
Bodamer OA, Leonard JV, Halliday D (1997) Dietary treatment in late-onset acid maltase deficiency. Eur J Pediatr 156(Supplement 1) S39–42.
Bodamer OA, Halliday D, Leonard JV (2000) The effects of l-alanine supplementation in late-onset glycogen storage disease type II. Neurology 55: 710–712.
Brown BI, Brown DH, Jeffrey PL (1970) Simultaneous absence of alpha-1,4-glucosidase and alpha-1,6-glucosidase activities (pH 4) in tissues of children with type II glycogen storage disease. Biochemistry 9(6): 1423–1428.
Curthoys NP, Watford M (1995) Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr 15: 133–159.
DiMauro S, Stern LZ, Mehler M, Nagle RB, Payne C (1978) Adult-onset acid maltase deficiency: a postmortem study. Muscle Nerve 1: 27–36.
Fingar DC, Blenis J (2004) Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 23: 3151–3171.
Hardie DG (2003) Minireview: The AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 144(12): 5179–5183.
Harris RA, Kobayashi R, Murakami T, Shimomura Y (2001) Regulation of branched-chain α-keto acid dehydrogenase kinase expression in rat liver. J Nutr 131: 841S–845S.
Harris RA, Joshi M, Jeoung NH, Obayashi M (2005) Overview of the molecular and biochemical basis of branched-chain amino acid catabolism. J Nutr 135(6 Supplement): 1527S–1530S.
Labow BI, Souba WW, Abcouwer SF (2001) Mechanisms governing the expression of the enzymes of glutamine metabolism—glutaminase and glutamine synthetase. J Nutr 131(9 Supplement): 2467S–2474S.
Macchi FD, Shen FJ, Keck RG, Harris RJ (2000) Amino acid analysis, using postcolumn ninhydrin detection, in a biotechnology laboratory. Methods Mol Biol 159: 9–30.
Mochel F, deLonlay P, Touati G, et al (2005) Pyruvate carboxylase deficiency: immediate clinical and biochemical improvement with dietary triheptanoin. Mol Gen Metab 84: 305–312.
Nehlig A, Pereira de Vasconcelos A (1993) Glucose and ketone utilization by the brain of neonatal rats. Prog Neurobiol 40(2): 163–221.
Rashed MS, Bucknall MP, Little D, et al (1997) Screening blood spots for inborn errors of metabolism by electrospray tandem mass spectrometry with a microplate batch process and a computer algorithm for automated flagging of abnormal profiles. Clin Chem 43: 1129–1141.
Roe CR, Sweetman L, Roe DS, David F, Brunengraber H (2002) Effective dietary treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Invest 110(2): 259–269.
Salway JG (2004) Metabolism at a Glance, 3rd edn. Oxford: Blackwell.
Saudubray JM, Martin D, De Lonlay P, et al (1999) Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis 22: 488–502.
Slonim AE, Coleman RA, McElligot MA, et al (1983) Improvement of muscle function in acid maltase deficiency by high-protein therapy. Neurology 33: 34–38.
Sweetman L (1991) Organic acid analysis. In: Hommes FA, ed. Techniques in Diagnostic Human Biochemical Genetics: A Laboratory Manual. New York: Wiley-Liss, 143–176.
Van der Walt JD, Swash M, Leake J, Cox EL (1987) The pattern of involvement of adult-onset acid maltase deficiency at autopsy. Muscle Nerve 10: 272–281.
Watford M (2000) Glutamine and glutamate metabolism across the liver sinusoid. J Nutr 130(4S Supplement): 983S–987S.
Watford M, Chellaraj V, Ismat A, Brown P, Raman P (2002) Hepatic glutamine metabolism. Nutrition 18(4): 301–303.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicating editor: Jean-Marie Saudubray
Competing interests: None declared
Rights and permissions
About this article
Cite this article
Roe, C.R., Mochel, F. Anaplerotic diet therapy in inherited metabolic disease: Therapeutic potential. J Inherit Metab Dis 29, 332–340 (2006). https://doi.org/10.1007/s10545-006-0290-3
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s10545-006-0290-3