
Abstract
The rapid expansion of multicellular native and alien species outbreaks in aquatic and terrestrial ecosystems (bioinvasions) may produce significant impacts on bacterial community dynamics and nutrient pathways with major ecological implications. In aquatic ecosystems, bioinvasions may cause adverse effects on the water quality resulting from changes in biological, chemical and physical properties linked to significant transformations of the microbial taxonomic and functional diversity. Here we used an effective and highly sensitive experimental strategy, bypassing the efficiency bottleneck of the traditional bacterial isolation and culturing method, to identify changes of the planktonic microbial community inhabiting a marine coastal lagoon (Varano, Adriatic Sea) under the influence of an outbreak-forming alien jellyfish species. Water samples were collected from two areas that differed in their level of confinement inside in the lagoon and jellyfish densities (W, up to 12.4 medusae m−3; E, up to 0.03 medusae m−3) to conduct a snapshot microbiome analysis by a metagenomic approach. After extraction of the genetic material in the environmental water samples, we deep-sequenced metagenomic amplicons of the V5–V6 region of the 16S rRNA bacterial gene by an Illumina MiSeq platform. Experiments were carried out in triplicates, so six libraries of dual indexed amplicons of 420 bp were successfully sequenced on the MiSeq platform using a 2 × 250 bp paired-end sequencing strategy. Approximately 7.5 million paired-end reads (i.e. 15 million total reads) were generated, with an average of 2.5 million reads (1.25 M pairs) per sample replicate. The sequence data, analyzed through a novel bioinformatics pipeline (BioMaS), showed that the structure of the resident bacterial community was significantly affected by the occurrence of jellyfish outbreaks. Clear qualitative and quantitative differences were found between the western and eastern areas (characterized by many or few jellyfish), with 84 families, 153 genera and 324 species in the W samples, and 104 families, 199 genera and 331 species in the E samples. Significant differences between the two sampling areas were particularly detected in the occurrence of 16 families, 22 genera and 61 species of microbial taxa. This is the first time that a NGS platform has been used to screen the impact of jellyfish bioinvasions on the aquatic microbiome, providing a preliminary assessment of jellyfish-driven changes of the functional and structural microbial biodiversity.
Similar content being viewed by others


Introduction
The recent advent of next generation sequencing technologies has revolutionized microbiome research, allowing unprecedented depth and resolution at affordable costs and, thus, enabling large-scale studies of microorganisms living in taxonomically and physically complex habitats. Because microbes have lived on Earth for more than 3 billion years, they are ubiquitous and play important roles in the Earth’s ecosystems (Kowalchuk et al. 2008). In particular, they participate in essential biogeochemical processes such as the carbon, nitrogen and sulphur cycles in terrestrial and aquatic ecosystems, are producers and decomposers in food webs (Dorigo et al. 2005; Kisand et al. 2012; Zehr 2010), and are primarily responsible for degradation of a large variety of natural organic compounds (Ogawa et al. 2001).
Aquatic environments harbor abundant and diverse microbial communities that ensure their functioning and sustainability (Azam and Malfatti 2007; Zinger et al. 2012). In recent decades, the importance of bacterioplankton has been persuasively demonstrated in nutrient cycling and food-web structure in the marine environment (Azam 1998; Cho 1990) and a multitude of marine bacteria have been isolated in ocean sites and different coastal areas of temperate, tropical and polar zones (Pommier et al. 2005; Rusch et al. 2007; Stabili and Cavallo 2011). Aquatic microbes are genetically, physiologically and ecologically diverse, and exhibit many different physiological responses, adaptation and evolutionary patterns (Bahgat 2011; Kemp and Aller 2004). The microbiota composition varies as a function of water class (e.g. oceans, lakes, rivers, springs, ponds and ground water) and it is affected by different factors such as water salinity, organic compound concentration, turbidity, temperature, and contamination sources (Bahgat 2011).
Study of the overall biodiversity is crucial for assessing and monitoring aquatic ecosystems; however, the composition, abundance, and distribution patterns of microbial communities usually remain poorly explored (Kisand et al. 2012; Zinger et al. 2012).
Previously, difficulties in collecting representative samples in particular habitats (e.g. deep sea) and experimental limitations related to the characterization of the uncultivable microbes have been the two main limitations for comprehensive assessments of the microbial communities living in certain environments (Kisand et al. 2012). This view recently changed with the development of culture-independent approaches, such as metagenomics. As defined by Thomas et al. (2012), metagenomics represent the direct genetic analysis of genomes contained in an environmental sample, without isolation or culture of individual organisms. The metagenomic approach is experiencing an explosive improvement since the advent of high-throughput Next-Generation Sequencing (NGS) technologies, which allow an unprecedented large-scale identification of organisms and communities through the production of an enormous amount of genetic data (Bourlat et al. 2013). Either of two experimental strategies can be applied starting from the total extracted DNA: (1) the entire metagenome is sequenced by a shotgun approach, or (2) gene markers specific for a given taxon are selectively sequenced after their amplification by PCR primer pairs able to function over a broad taxonomic range (amplicon-based approach). The second approach is the most common strategy applied in various ecological settings and is used to investigate prokaryotic diversity through analysis of hypervariable regions of the 16S rRNA gene (Hajibabaei et al. 2011; Klindworth et al. 2013). Illumina/Solexa represents the NGS sequencing system most widely used for amplicon-based metagenomic studies (Luo et al. 2012), providing optimal performances in terms of time, costs and coverage (Caporaso et al. 2012; Kozich et al. 2013; Liu et al. 2012; Loman et al. 2012; Quail et al. 2012). In particular, the Illumina MiSeq benchtop sequencer is very suitable for amplicon-based metagenomic studies, considering its overall features including the obtainable read length up to 300 m. Nevertheless, few available experimental protocols for the amplicon-based metagenomic approach on the MiSeq platform are available involving the application of the TruSeq (Caporaso et al. 2012; Gilbert et al. 2010; Rubin et al. 2013) or a custom designed approach (Kozich et al. 2013).
We used a flexible strategy including both a NGS library preparation based on the Illumina-Nextera protocol and a user-friendly data analysis workflow (BioMaS, Bioinformatic analysis of Metagenomic AmpliconS) for monitoring changes in the planktonic microbiome potentially related to the invasion and proliferation of a non-indigenous jellyfish population (the moon jelly Aurelia sp.) in a lagoonal habitat, the Varano lagoon (South Adriatic). This strategy may allow the production of a large number of high quality sequencing reads, minimizing the cost per run and simplifying the experimental procedure.
Jellyfish blooms are occurring with increasing frequency and magnitude in many estuarine and coastal habitats (Attrill et al. 2007; Boero et al. 2008; Mills 2001; Purcell 2012). In the Mediterranean Sea, Aurelia sp. invasions are widely recorded, in the Mljet lakes, Croatia (Malej et al. 2007), in Etang de Thau, France (Bonnet et al. 2012), in the Gulf of Trieste, Italy and Slovenia (Malej et al. 2012). This is also the case for the Varano lagoon, a coastal lagoonal habitat with a strong aquaculture vocation and tradition. The Varano ecosystem was invaded by the moon jellyfish Aurelia sp. more than 10 years ago and now has a persistent population with high densities in some parts (Belmonte et al. 2011; Manini et al. 2005; Scorrano 2014).
The invasion of Aurelia jellyfish in coastal lagoons often can be related to the introduction of mussel culture rafts bearing the polyp stages, which asexually release great numbers of medusae into the water column (Lo et al. 2008; Purcell, 2012). In Varano lagoon, eventually a year-round resident population was established with high densities of jellyfish (mean 4.5 ind) in spring and summer months. Molecular COI barcoding assigned the Varano jellyfish population to none of the presently known Aurelia species in the Mediterranean Sea (Aurelia sp. 5 and Aurelia sp. 8 sensu Dawson), suggesting a new jellyfish bioinvasion event (Scorrano, 2014). Mussel rafts are concentrated in the western part of Varano lagoon and may have introduced attached jellyfish polyps to the lagoon. Active swimming of the jellyfish against the main W–E water flow may contribute to maintaining a spatial segregation of jellyfish in the western part of the lagoon.
Jellyfish may acquire key roles in planktonic food webs by converting large quantities of carbon (C) into gelatinous biomass available only to a few higher consumers (Arai, 2005). Furthermore, the biomass of jellyfish generates a large amount of carbon-rich colloidal and dissolved organic matter (jelly-DOM) (Hansson and Norrman 1995) that is released into the water and easily available to the bacterioplankton (Carlson et al. 2002; Condon and Steinberg 2008), with consequences for bacterial growth and enzymatic activities (Riemann et al. 2006; Titelman et al. 2006). The protein-rich decay jellyfish biomass and the mucous excretion of living jellyfish also are readily assimilated by jellyfish-associated and free-living bacteria and can cause rapid shifts in microbiome structure and organization (Tinta et al. 2010, 2012). The bacterial use of jelly-DOM could lead to the repackaging of gelatinous organic carbon and its reincorporation into the food web (Condon et al. 2011). In addition, the decay of gelatinous biomass (jellyfalls) following bloom events leads to the rapid and massive release in the water column of dissolved inorganic and organic compounds (Pitt et al. 2009).
The present investigation focused on assessing the potential of NGS-based methods for rapid detection of structural and functional changes in the microbial community associated with jellyfish blooms in a marine coastal lagoon.
Materials and methods
Sampling
The Varano lagoon is a shallow enclosed coastal basin (Fig. 1) located on the southern Adriatic coast (Apulia region, Italy) on the north side of the Gargano promontory. The lagoon has a 33 km perimeter and covers an area of over 60.5 km2. The lagoon represents a typical eurythermal habitat (low 7 °C, high up to 30 °C), with a maximum depth of 5 m and high inter-seasonal physico-chemical variability. Freshwater springs are distributed all around the lagoon perimeter (Fig. 1) and two artificial channels (foce Capojale, NW; foce Varano, NE) regulate lagoon-sea water exchanges (Specchiulli et al. 2008), facilitating the hydrodynamic balance of sea water masses at reduced salinity (25–29 ppt) through tidal and wind forces. Prevailing N–NE winds make these exchanges much more effective in autumn and winter (Spagnoli et al. 2002), with the main sea water inflow through the NW channel. Due to the small tidal excursion and reduced exchange with the adjacent coastal area, water residence time is very long (about 1.5 years; Specchiulli et al. 2008).

Map of Varano lagoon (geographical coordinates on the axes) with West (W) and East (E) replicate sampling points (plus symbol) and fresh water springs (asterisk)
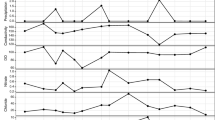
To reveal changes in the planktonic bacterial communities in the lagoon relative to the direct or indirect impact of the jellyfish biomass, a metagenomic approach was applied to triplicate water samples collected on 17 July 2013 in two areas of the lagoon (Fig. 1), designated West (W) and East (E), and differing in their level of confinement (Melaku Canu et al. 2012) and jellyfish densities (W: up to 12.4 ind m−3; E: up to 0.03 ind m−3; Scorrano 2014). Samples were collected in pre-sterilized bottles at −0.50 m depth and stored at +4 °C until filtration through a 0.2-μm pore size filter (47 mm diameter) MO BIO (MoBio Laboratories, Inc. CA, USA). Sample membranes then were stored at +4 °C until DNA extraction, which was carried out within 4 h from the sampling. Vertical profiles of temperature, salinity, pH, depth, and dissolved oxygen were recorded every 6 weeks by a multiparametric CTD probe Ocean Seven 401 (IDRONAUT; Scorrano 2014). From that dataset, we obtained the means ± standard deviations of temperature, salinity, and oxygen saturation at 0.5 m depth in triplicate samples collected at W and E areas on the day of water sample collection.
DNA extraction
Microbial DNA was extracted from each filter (3 replicates/area) using the MO BIO PowerWater® DNA Isolation Kit (MoBio Laboratories) following the manufacturer’s instructions. The bead-beating step was performed in the FastPrep Instrument (BIO 101, Carlsbad, Canada) for 40 s at speed 6. The homogenized samples were centrifuged (4,000×g for 3 min at room temperature) and the supernatant subjected to the subsequent protein and inhibitor removal steps as recommended by the manufacturer. DNA was eluted in 100 μl of the PW6 solution. The quantity and quality of the extracted DNA were assessed by spectrophotometric quantification using NanoDrop 2000c (Thermo Fisher Scientific, Inc., DE, USA) and agarose gel (1.2 %) electrophoresis, respectively. The extracted DNA was stored at −20 °C.
Library preparation and sequencing
Among the nine hypervariable regions present in the 16S rRNA gene, the V5–V6 regions were chosen as the target for prokaryotic identification (Chakravorty et al. 2007). The microbial DNA extracted from each sample, named respectively W1, W2, W3 and E1, E2, E3, was used as templates for the amplicon preparation. The strategy adopted for the amplicon library preparation consisted of two successive rounds of PCR. In the first round, the V5–V6 regions were amplified from the extracted DNA using the overhang primer pairs, named V5–V6 NextFor/Rev (5′TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG[ATTAGATACCCYGGTAGTCC]3′/5′GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG[ACGAGCTGACGACARCCATG]3′) and designed to contain (from 5′ to 3′ends) the transposon Nextera’s sequences (Nextera DNA sample preparation guide, Illumina) and the universal BV5 (Next For) and AV6 (Next Rev; Stecher et al. 2010) priming sequences (underlined nucleotides).
Amplification was performed using the Phusion® High-Fidelity DNA polymerase (Thermo Fisher Scientific, Inc., New England Biolabs) in a Mastercycler Thermal Cycler (Eppendorf, Hamburg, Germany). Each reaction mixture contained 0.25 ng of extracted DNA, 1X Buffer HF, 0.2 mM dNTPs, 0.5 μM of each primer, and 1U Phusion DNA Polymerase in a final volume of 50 µl. The cycling parameters for PCR were standardized as follows: initial denaturation 98 °C for 30 s, followed by 10 cycles of denaturation at 98 °C for 10 s, annealing at 58 °C for 30 s, extension at 72 °C for 15 s, and subsequently 15 cycles of denaturation at 98 °C for 10 s, annealing at 62 °C for 30 s, extension at 72 °C for 15 s, with a final extension step of 7 min at 72 °C. All PCRs were performed in the presence of a negative (Molecular Biology Grade Water, RNase/DNase-free water) and a positive (genomic DNA from pure culture of Oenococcus oeni) control. The PCR products (around 354 bp long) were visualized on 1.2 % agarose gel and purified using the AMPure XP Beads at a concentration of 0.8× vol/vol (Agencourt Bioscience Corporation, Beverly, Massachusetts).
The purified amplicons were used as templates in the second PCR round, which was performed with the Nextera indices priming sequences as required by the dual index approach reported in the Nextera DNA sample preparation guide (Illumina). The dual index strategy consists of incorporating unique indices into both ends of the library molecules to allow sample identification for the subsequent bioinformatics analysis (Kozich et al. 2013).
The 50 μl reaction mixture was made up of the following reagents: template DNA (40 ng), 1X Buffer HF, dNTPs (0.1 mM), Nextera index primers (index 1 and 2) and 1U Phusion DNA Polymerase system. The cycling parameters were those suggested by the Illumina Nextera protocol. The dual indexed amplicons obtained (~420 bp) were purified using magnetic beads AMPure XP (0.6X, vol/vol), checked for quality control on Bioanalyzer 2100 (Agilent Technologies, Santa Clara, California) and quantified by fluorimetry using the Quant-iTTM PicoGreen® dsDNA Assay Kit (Invitrogen, Carlsbad, California). Finally, equimolar ratios of the purified amplicons (six in total) were pooled and subjected to 2 × 250 bp paired-end sequencing on the MiSeq platform. In order to increase the genetic diversity, as required by the MiSeq platform, the phage PhiX genomic DNA library was added to the mix and co-sequenced (Kozich et al. 2013).
Taxonomic binning
A modular pipeline, called BioMaS developed in our laboratory, was applied to analyze the paired-end (PE) reads obtained by the six samples analyzed (3 replicates/area) in order to characterize the bacterial composition. Before BioMaS analysis, a statistical and quality check of the reads was carried out by FastQC and denoising performed by trim-galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) setting a quality-score threshold equal to 25.
The BioMaS pipeline consists mainly of three steps. Because most of the generated reads were overlapping due to the amplicon length (~420 bp) being shorter than the total sequenced PE length (2 × 250 bp), in the first step, the overlapping PE reads were merged into contiguous consensus sequences using Flash (Magoc and Salzberg 2011). These were subsequently dereplicated by Usearch (Edgar 2010), but retaining the information about the total number of original consensus sequences. A small proportion of PE reads (generally about 5 %), which did not overlap to form a consensus contig probably due to the sequencing of amplicon subfragments. These were further cleaned through the removal of low quality regions and retained for further analysis as paired-end if both sequenced reads were ≥50 bp long; otherwise, they were discarded. Then, a similarity-based analysis was performed in order to infer the taxonomic origin of the generated metagenomic sequences. In particular, the consensus sequences and the non-overlapping pairs were mapped against the RDP database (Ribosomal Database Project; release 11.2; Cole et al. 2009) by means of Bowtie2 (Langmead and Salzberg 2012) and the mapping data were filtered using a Python script according to two parameters: identity % (≥97 %) and query coverage (≥95 %). The choice of this stringent % identity threshold allows classifying most of the reads up the species level (Mende et al. 2013). Finally, all mapped reads fulfilling the filters set were taxonomically annotated using the Tango tool (Clemente et al. 2011). All the residual unclassified reads were clustered in UTU (unknown taxonomic units) at ≥97 % similarity by Usearch and those represented by fewer than 5 sequences in all samples were discarded. Statistically significant differences of the bacterial population in different samples were determined at family, genus and species levels using the DESeq, an R/Bioconductor package (Anders and Huber 2010).
Results
The water samples among the low-jellyfish (E1, E2, E3) and high-jellyfish (W1, W2, W3) areas did not differ significantly in the measured abiotic parameters (T, S, oxygen saturation; Table 1), merely showing a slight intrusion of cold freshwater in the E zone. Differences in oxygen saturation were not significant between the two sectors. From the membrane filters, microbial DNA samples (W1, W2, W3, E1, E2, E3) were extracted with average DNA yields of 2.64 and 1.97 μg for W and E samples, respectively.
Six libraries of dual indexed amplicons of 420 bp (Fig. 2) were successfully sequenced on the MiSeq platform using a 2 × 250 bp paired-end sequencing strategy. Approximately 7.5 million paired-end (PE) reads (i.e. 15 million total reads) were generated, averaging 1.25 million PE reads (2.5 M reads) per sample replicate (see Sup. Table 1 in Supplementary Material). All sequenced samples generated reads of high quality, mostly having the expected length of 250 bp. The paired-end reads produced for the six samples (3 replicates/area) were subjected to BioMaS computation. First, the paired-end reads were merged and low quality regions removed from the non-overlapping reads. About 94 % of the paired-end reads were merged into a consensus sequence, while 2 % were treated as paired-end and the remaining 4 % were discarded. The sequences passing the quality control were then compared to the RDP database content. About 86.6 % of the analyzed sequences had at least one match in RDP above the adopted similarity cut-off (97 %). Finally, the taxonomic binning of the matching reads was carried out by using TANGO (Clemente et al. 2011). The PE reads matching against RDP with a percent identity above the threshold (86.6 %) were classified at the family (74.2 %), genus (38.2 %) and species (8.6 %) levels (Supplementary Tables). Remarkably, rarefaction curves (Fig. S1) indicated that species richness was comprehensively represented in all samples.

Agilent Bioanalyzer chip profile of purified amplicons (420 bp), relative to V5–V6 hypervariable region of the bacterial 16S rRNA gene, obtained applying the modified Nextera’s protocol for the six samples (W1, W2, W3 and E1, E2, E3). L, molecular weight marker. Lanes 1–3, West samples (W1, W2, W3); lanes 4–6, East samples (E1, E2, E3). Bands at 35 and 10,380 bp represent internal size standards and not DNA from the sample
A total of 109 families, 213 genera and 353 species were represented in the samples, with clear differences between the W samples with high jellyfish density (84 families, 153 genera and 324 species) and the E samples with low-no jellyfish (Scorrano 2014; 103 families, 199 genera and 331 species; Supplementary Material).
The heat map showed clear-cut clustering of the bacterial populations in W and E samples (Fig. 3). Furthermore, the beta-diversity values calculated for all nine pairwise comparisons involving the W and E samples supported very good reproducibility between microbial communities among the three replicates (0.151 ± 0.016).

Heat map visualization at the family level of the six samples (W1, W2, W3 and E1, E2, E3), obtained applying the DESeq package on the taxonomic data produced by BioMaS, which show the clustering of samples from jellyfish-rich and jellyfish-poor sites
Statistically significant differences between the aquatic bacterial communities in the W and E zones were determined at family, genus and species levels using DESeq (Table 2 and Supplementary Tables 3, 5, 7). In particular, we observed significant differences in the occurrence of 16 families (Halomonadaceae, Saprospiraceae, Shewanellaceae, Flavobacteriaceae, Chloroplast, Oceanospirillaceae, Cryomorphaceae, Comamonadaceae, Flammeovirgaceae, Rhodobacteraceae, Family_II (Cyanobacteria), Acidimicrobiaceae, Simkaniaceae, Acholeplasmataceae, Rhodospirillaceae, Microbacteriaceae), 22 genera and 61 species in the W and E samples (Table 2).
The families Halomonadaceae, Saprospiraceae, Shewanellaceae, Chloroplast, Flammeovirgaceae, Family_II (Cyanobacteria), and Simkaniaceae were significantly enriched in the jellyfish-rich (W) zone with an over-representation of several genera including Halomonas, Haliscomenobacter, Shewanella, Fabibacter, Roseivirga and Owenweeksia (see the relevant normalized counts in Supplementary Material). Interestingly, the amplicon based approach detected also the chloroplast rRNA of Bacillariophyta and an over-representation of diatoms the W zone was significantly supported. Conversely, the families Oceanospirillaceae, Comamonadaceae, Acholeplasmataceae, Acidimicrobiaceae, Cryomorphaceae, Flavobacteriaceae, Rhodobacteraceae, Rhodospirillaceae, Microbacteriaceae were significantly enriched in the jellyfish-poor (E) zone with several over-represented genera including Cellulophaga, Crocinitomix, Ilumatobacter, Diaphorobacter. The tree in Fig. S2 graphically illustrates significantly over- and under- represented families and genera in the jellyfish-rich zones (all six sample-specific trees generated by BioMaS tool are available in Supplementary Figs. 3–8).
About 12.8 % of the reads did not show any significant match (≥97 % identity) against the RDB. Those reads were probably derived from novel species not represented in the RDB and were classified as UTU. In total, we detected 44 UTUs, showing significantly different abundances in the W and E zones (Supplementary Tables 8–9).
Discussion
Molecular tools are increasingly used for screening biodiversity in terrestrial and aquatic ecosystems to develop appropriate management programs (e.g. Bourlat et al. 2013; Darling and Mahon 2011; Geller et al. 2010; Karp et al. 1997). In the marine environment, molecular methods can reveal patterns of community dynamics and ecosystem functioning processes by rapid detection of the identities of a vast majority of taxa in a community and, therefore, are used for environmental monitoring programs, directly contributing to development of management strategies of coastal areas. Molecular methods can play an important role in several fields of aquatic bioinvasions, including identification of non-indigenous species and their origins, historical reconstruction of invasive routes and the underlying pathways, estimation of genetic connectivity among populations, as well as detection of overall changes in the residential native community induced by new invaders (e.g. Darling and Blum 2007; Darling and Mahon 2011; Estoup and Guillemaud 2010; Geller et al. 2010; Ghabooli et al. 2013; Hänfling 2007; Miura 2007; Muirhead et al. 2008; Piraino et al. 2014). In particular, NGS platforms are known as rapid and sensitive means of characterizing complex microbial communities, and are supplanting traditional surveys of microbial diversity based on initial cultivation screenings followed by PCR amplification, cloning and sequencing of universally conserved molecules, such as the 16S rRNA gene (reviewed by Degnan and Ochman 2012).
We investigated the changes in the planktonic microbiome diversity of a lagoon habitat probably driven by the invasion and proliferation of a non-indigenous jellyfish (Aurelia sp.). In parallel, this study offered the opportunity to verify the effectiveness of coupling the Illumina protocol with the BioMaS pipeline. Significant differences in microbiota abundance were determined at family, genus and species levels (Table 2 and Supplementary Material) and interpreted as related to changes in the nutrient availability. Indeed, jellyfish carcasses and mucous exudates possibly serve as important carbon sources for these bacteria. The total organic content of jellyfish, in fact, generally consists of carbohydrates (7 ± 5 %), intermediate lipids (22 ± 12 %), and especially proteins (72 ± 14 %; Billett et al. 2006; Doyle et al. 2006; Pitt et al. 2009; Yamamoto et al. 2008).
Interestingly, some of these results are confirmed by a study on the microbial diversity in the Varano lagoon conducted by traditional analysis of the cultivable heterotrophic bacterial community, followed by the standard PCR-based amplification of the 16S rRNA gene (Caprioli 2014; Stabili et al. 2013). This study from cultivable bacteria corroborated a significant difference between low-jellyfish and high-jellyfish areas of the lagoon, and showed a prevalence of Shewanellaceae in the presence of jellyfish, as also detected by our metagenomic approach. Overall, jellyfish may influence bacterial abundance and composition by shifting the native microbial community towards more opportunistic consortia with higher metabolic potential for exploitation of mucopolysaccharides and glycoproteins from jellyfish tissues. In spite of considerable evidence for predation on jellyfish tissues (e.g. Arai 2005; Milisenda et al. 2014), jellyfish are usually considered to be a low-value biomass not easily available to higher trophic levels and dependent on the microbial loop for recycling (Condon et al. 2011). Overall, the observed differences in the bacterial community might be interpreted as an ecosystem response to allow adaptation of metabolic pathways to the jellyfish-derived organic carbon substrate. The biochemical and ecological characteristics of the heterotrophic bacterial taxa that are selectively promoted in the Varano lagoon in the areas under massive jellyfish blooms reveal some adaptive features favoring microbial growth and selection on jellyfish tissues or on decaying jellyfall organic matter, as follows.
The phenotypically diverse family Halomonadaceae consists mostly of marine and moderately halophilic bacteria (Arahal et al. 2008) adapted to a wide range of salinities. Members of this family are Gram-negative, either slight or moderate halophiles with straight or curved, rod-shaped cells (Baumann et al. 1983; Bouchotroch et al. 2001; Duckworth et al. 2000; Franzmann et al. 1987; Mellado et al. 1995; Mormile et al. 1999; Quesada et al. 1984, 1990; Valderrama et al. 1991; Vreeland et al. 1980). This bacterial group is characterized by the production of extracellular polymers including exopolysaccharides (EPS), which has been described as a strategy for growth (Costerton 1999). Studies of bacteria growing in aquatic systems on marine sediments, aggregates and detrital particles show that nearly all of the cells are surrounded by EPS (Costerton 1999; Decho 1990) and many of the cells are enclosed with adherent biofilms. Thus, the significantly higher abundance of Halomonadaceae in the W zone might be related to their ability to produce EPS for adhesion to rich particulate organic matter derived from jellyfall aggregates. As an example, Halomonas rifensis is a recently described species of the genus (Amjres et al. 2011). It is an EPS-producing, halophilic bacterium capable of growth in salt concentrations (mixture of sea salts) of 0.5–20 % (weight/volume, optimum 5–7.5 % w/v), incapable of growing without NaCl, grows at 25–45 °C (optimum 32 °C) and at pH 5–10 (optimum pH 6–9). Nevertheless, the jellyfish-associated growth of Halomonadaceae might be an indirect consequence due to release from competition with less tolerant prokaryotic groups. Interestingly, also in Jellyfish Lake, Palau known for the large resident population of Mastigias papua, members of Halomonodaceae dominated among cultivable heterotrophic bacteria (Venkateswaran et al. 1993).
The family Shewanellaceae was established from the amended description of a group of marine Alteromonas-like bacteria (Ivanova et al. 2004a, b, c). The Shewanellaceae comprises Gram-negative, straight or curved rod-shaped, aerobic or facultative anaerobic and readily cultivated gammaproteobacteria isolated frequently in the marine environment from diverse sources, including red algae (Simidu et al. 1990), a tidal flat (Yoon et al. 2004a), seawater (Ivanova et al. 2001, 2004b; Yoon et al. 2004b), sediments (Venkateswaran et al. 1998) and marine invertebrates (Ivanova et al. 2004c). In particular, Shewanella spp., significantly abundant in the W zone, have been recovered from red algae and are occasionally human pathogens (Sharma and Kalawat 2010).
Although bacteria of the genus Shewanella belong to one of the readily cultivable groups of microorganisms, little is known about their occurrence and abundance in marine ecosystems. Members of Shewanella usually are found in marine environments in warm climates or during summer in temperate climates. One of the striking features of these bacteria is the ability of some recently described species to produce polyunsaturated fatty acids (PUFAs) at relatively high incubation temperatures (25−30 °C; Ivanova et al. Ivanova et al. 2001, 2003; Skerratt et al. 2002), which contradicts the idea that only barophilic or cold-adapted species are able to produce significant levels of PUFAs. Notably, most of these microorganisms, including several Shewanella species particularly abundant in the W zone, are metabolically active and produce a range of hydrolytic enzymes, i.e. proteinases (gelatinases and caseinases), lipases, amylases, agarases, and alginases. It is most likely that bacteria of this genus can actively degrade organic matter, including the gelatinous material from jellyfalls. It is noteworthy that the genus Shewanella is part of the normal bacterial symbiotic flora in cnidarians, including the moon jellyfish Aurelia aurita in the North Sea (Schuett and Doepke 2010; LaDouceur et al. 2013).
The families Flammeovirgaceae and Saprospiraceae, which were more abundant in the W zone, belong to the phylum Bacteroidetes also known as the Bacteroides–Cytophaga–Flexibacter group. Members of the phylum Bacteroidetes are the most abundant bacteria in the ocean after Proteobacteria and Cyanobacteria (Amaral-Zettler et al. 2011; Glockner et al. 1999; Kirchman 2002). They account for a significant fraction of marine bacterioplankton especially in coastal areas, where they represent between 10 and 30 % of the total bacterial counts (Alonso-Saez and Gasol 2007). Bacteroidetes are increasingly regarded as specialists in degradation of high molecular weight organic matter, i.e., proteins and carbohydrates (Thomas et al. 2011) and have a preference for growth attached to particles and surfaces, including algal cells. Because of these features, their significant presence in the W zone may be related to a lifestyle as jellyfish-associated bacteria. In particular, the family Flammeovirgaceae currently includes the genera Aureibacter, Csiribacter, Fabibacter, Flammeovirga, Flexithrix, Fulvivirga, Limibacter. Marivirga, Nafulsella, Perexilibacter, Persicobacter, Porifericola, Rapidithrix, Reichenbachiella Roseivirga, Sediminitomix and Tunicatimonas (Kim et al. 2013). Interestingly, among the genera determined by DESeq, Fabibacter and Roseivirga were significantly more abundant in the W zone. These two genera share many phenotypic features and were previously isolated together from the marine sponge Tedania ignis in the Bahamas (Lau et al. 2006). The family Saprospiraceae (Yoon et al. 2012) consists of the genera Aureispira, Haliscomenobacter, Lewinella, and Saprospira found in various habitats especially as epiphytic protein-hydrolyzing bacteria. Large, filamentous microorganisms are widely distributed in freshwater lakes (Schauer and Hahn 2005) and are believed to play an important roles there. Molecular evidence demonstrates that members of the family Saprospiraceae are also present in hypersaline mats (Ley et al. 2006), and three strains of gliding bacteria belonging to the genus Saprospira have been isolated from marine sponges and algae from the southern coastline of Thailand (Hosoya et al. 2006). Of these genera, only the genus Haliscomenobacter was detected in the W zone. Members of Bacteroidetes are generally associated with the degradation of complex organic materials (Cottrell and Kirchman 2000; Reichenbach 1991; Riemann et al. 2000), but other than Haliscomenobacter, little is known about the detailed ecophysiology of other Saprospiraceae. Isolates of Haliscomenobacter hydrolyze starch and grow aerobically on glucose, N-acetylglucosamine, lactose, and sucrose, but not on glycerol, lactate, acetate, and succinate (van Veen et al. 1973). Therefore, in the Varano lagoon in presence of jellyfish bloom, members of Haliscomenobacter may be involved in the hydrolysis of polysaccharides to utilize the hydrolysates as energy and carbon sources for growth. Thus, Bacteroidetes probably have a different life strategy that do other marine bacteria, such as Cyanobacteria (Family_II GpIIa), which are photoautotrophs and also were more abundant in the W zone.
The Micrococcaceae family is composed of bacteria with either respiratory and/or fermentative metabolism, using carbohydrate and/or amino acid substrates, which are the main components of jellyfish organic matter. In particular, Micrococcus, which was significantly more abundant in the W zone, includes ubiquitous Gram-positive bacteria species. Its presence in marine habitats has not been frequently reported, but unspecified micrococci were described from different seawater locations (Sieburth 1979), and a Micrococcus sp. was reported as a commensal of the sponge Tedania ignis (Stierle et al. 1988).
Few methodologies for Illumina-based analyses of microbial communities have been reported to date (Caporaso et al. 2012; Degnan and Ochman 2012; Kozich et al. 2013). In particular, the strategy of Caporaso et al. (2012) requires the synthesis of a large collection of different primers as a function of the number of samples to be tested; it was applied to analysis of only the variable V4 region of the 16S rRNA gene and referred to a previous single indexed Illumina sequencing system. More recently, Kozich et al. (2013) set up a dual index paired-read approach based on custom primers, employing a lower number of primer pairs during library construction. Nevertheless, in both approaches, the number of custom primers depends on the number of samples and especially on the number of regions to be tested in the same run. The strategy adopted here, based on the Nextera Illumina protocol and a dual-indexing principle, may overcome those issues, because it reduces the number of primer pairs to be used and, therefore, allows analysis of more samples per run at reduced costs.
The above results show significant differences in the composition of the community of bacteria and a few microalgae, between the samples from the W and E areas, separated by only 5–6 km, but characterized by high numbers or near absence of jellyfish, respectively. The simple experimental design of this pilot study specifically addressed the issue of the usefulness and reproducibility of NGS methods to rapidly obtain point information on ecological phenomena, such as jellyfish outbreaks, occurring within temporally limited windows. For this reason, we cannot rule out the possibility that the observed differences in the microbial structure in the water column might have preceded the jellyfish invasion, depending on area-specific, chemico-physical features and/or some biotic factors (e.g. organic matter, Chl-a content) and processes related to the structure and function of the biological communities (e.g. primary production, predation, competition, decomposition). Nonetheless, in spite of strong seasonal fluctuations of most environmental variables (as typical of confined coastal habitats), the Varano lagoon is apparently a relatively homogeneous basin from spatial, hydrological, and geomorphological standpoints. Previous studies on the water column, sediments, and biological features (organic matter, Chl-a, phyto- and zooplankton compositions) did not show significant differences across the lagoon and, specifically, between the two areas (high-jellyfish vs low-jellyfish) (Caroppo 2000; Spagnoli et al. 2002; Specchiulli et al. 2008), which are marked by a comparable level of confinement (e.g. in terms of relative distance from the open sea). One remarkable difference is the abundance of artificial structures in the western area of the lagoon, where mussel seed farming is common. Artificial substrates are usually known as the preferential site of settlement of planula larvae and their development into the polyp stage (Holst and Jarms 2007; Lo et al. 2008). The jellyfish spatial segregation might also be favoured by the jellyfish aggregative locomotion, actively directed against the water flow (Rakow and Graham 2006). The lack of consistent data corroborating alternative explanations, and the relative spatial homogeneity of the Varano lagoon both indirectly suggest that jellyfish outbreaks can be considered at least one of the main drivers of the microbial community differences between the two areas.
To our knowledge, this is the first time that a NGS platform has been used to screen changes of the aquatic microbiome associated with the proliferation of an invasive alien species. Here we valuated the potential of NGS analysis for assessing the impact of jellyfish outbreaks on carbon flow in aquatic food webs and ecosystem functioning, promisingly offering additional perspectives for the use of NGS methods in ecological investigations. Invasive jellyfish species producing large outbreaks in coastal ecosystems may have significant impacts on bacterial population dynamics and nutrient pathways with major ecological implications. The use of fast and sensitive methodologies, such as Illumina-based sequencing surveys, will allow rapidly gaining major insights into the functioning of coastal ecosystems and adopting the best practices for management and valuation of coastal habitats and their living resources.
References
Alonso-Saez L, Gasol JM (2007) Seasonal variations in the contributions of different bacterial groups to the uptake of low-molecular-weight compounds in northwestern Mediterranean coastal waters. Appl Environ Microbiol 73(11):3528–3535. doi:10.1128/AEM.02627-06
Amaral-Zettler LA, Zettler ER, Theroux SM, Palacios C, Aguilera A, Amils R (2011) Microbial community structure across the tree of life in the extreme Rio Tinto. ISME J 5(1):42–50. doi:10.1038/Ismej.2010.101
Amjres H, Bejar V, Quesada E, Abrini J, Llamas I (2011) Halomonas rifensis sp nov., an exopolysaccharide-producing, halophilic bacterium isolated from a solar saltern. Int J Syst Evol Microbiol 61:2600–2605. doi:10.1099/Ijs.0.027268-0
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11(10):R106. doi:10.1186/gb-2010-11-10-r106
Arahal DR, Vreeland RH, Litchfield CD, Mormile MR, Tindall BJ, Oren A, Bejar V, Quesada E, Ventosa A (2008) Recommended minimal standards for describing new taxa of the family Halomonadaceae (vol 57, pg 2436, 2007). Int J Syst Evol Microbiol 58:2673
Arai MN (2005) Predation on pelagic coelenterates: a review. J Mar Biol Assoc UK 85(3):523–536
Attrill MJ, Wright J, Edwards M (2007) Climate-related increases in jellyfish frequency suggest a more gelatinous future for the North Sea. Limnol Oceanogr 52(1):480–485
Azam F (1998) Microbial control of oceanic carbon flux: the plot thickens. Science 280(5364):694–696. doi:10.1126/science.280.5364.694
Azam F, Malfatti F (2007) Microbial structuring of marine ecosystems. Nat Rev Microbiol 5(10):782–791. doi:10.1038/nrmicro1747
Bahgat M (2011) Diversity of bacterial communities in contrasting aquatic environments: Lake Timsah, Egypt. Microbiol Insights 4:11–19. doi:10.4137/mbi.s6948
Baumann L, Bowditch RD, Baumann P (1983) Description of Deleya gen. nov. created to accommodate the marine species Alcaligenes aestus, A. pacificus, A. cupidus, A. venustus, and Pseudomonas marina. Int J Syst Bacteriol 33(4):793–802. doi:10.1099/00207713-33-4-793
Belmonte G, Scirocco T, Denitto F (2011) Zooplankton composition in Lake Varano (Adriatic Sea coast, Italy). Ital J Zool 78(3):370–378. doi:10.1080/11250003.2011.561261
Billett DSM, Bett BJ, Jacobs CL, Rouse IP, Wigham BD (2006) Mass deposition of jellyfish in the deep Arabian Sea. Limnol Oceanogr 51(5):2077–2083
Boero F, Bouillon J, Gravili C, Miglietta MP, Parsons T, Piraino S (2008) Gelatinous plankton: irregularities rule the world (sometimes). Mar Ecol Prog Ser 356:299–310. doi:10.3354/meps07368
Bonnet D, Molinero JC, Schohn T, Yahia MND (2012) Seasonal changes in the population dynamics of Aurelia aurita in Thau lagoon. Cah Biol Mar 53(3):343–347
Bouchotroch S, Quesada E, del Moral A, Llamas I, Bejar V (2001) Halomonas maura sp nov., a novel moderately halophilic, exopolysaccharide-producing bacterium. Int J Syst Evol Microbiol 51:1625–1632
Bourlat SJ, Borja A, Gilbert J, Taylor MI, Davies N, Weisberg SB, Griffith JF, Lettieri T, Field D, Benzie J, Glockner FO, Rodriguez-Ezpeleta N, Faith DP, Bean TP, Obst M (2013) Genomics in marine monitoring: new opportunities for assessing marine health status. Mar Pollut Bull 74(1):19–31. doi:10.1016/j.marpolbul.2013.05.042
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6(8):1621–1624. doi:10.1038/ismej.2012.8
Caprioli R (2014) Influence of jellyfish biomass on bacterial dynamics in the lake of Varano. PhD Thesis, University of Modena e Reggio Emilia, Modena
Carlson CA, Giovannoni SJ, Hansell DA, Goldberg SJ, Parsons R, Otero MP, Vergin K, Wheeler BR (2002) Effect of nutrient amendments on bacterioplankton production, community structure, and DOC utilization in the northwestern Sargasso Sea. Aquat Microb Ecol 30(1):19–36. doi:10.3354/Ame030019
Caroppo C (2000) The contribution of picophytoplankton to community structure in a Mediterranean brackish environment. J Plankton Res 22(2):381–397. doi:10.1093/Plankt/22.2.381
Chakravorty S, Helb D, Burday M, Connell N, Alland D (2007) A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J Microbiol Methods 69(2):330–339. doi:10.1016/j.mimet.2007.02.005
Cho BC (1990) Biogeochemical significance of bacterial biomass in the ocean’s euphotic zone. Mar Ecol Prog Ser 63(2–3):253–259. doi:10.3354/meps063253
Clemente JC, Jansson J, Valiente G (2011) Flexible taxonomic assignment of ambiguous sequencing reads. BMC Bioinformatics 12:8. doi:10.1186/1471-2105-12-8
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37(Database issue):D141–D145. doi:10.1093/nar/gkn879
Condon R, Steinberg D (2008) Development, biological regulation, and fate of ctenophore (Mnemiopsis leidyi) blooms in the York River estuary, USA. Mar Ecol Prog Ser 369:153–168
Condon RH, Steinberg DK, del Giorgio PA, Bouvier TC, Bronk DA, Graham WM, Ducklow HW (2011) Jellyfish blooms result in a major microbial respiratory sink of carbon in marine systems. Proc Natl Acad Sci USA 108(25):10225–10230. doi:10.1073/Pnas.1015782108
Costerton JW (1999) The role of bacterial exopolysaccharides in nature and disease (Reprinted from Developments in Industrial Microbiology, vol 26, pg 249-261, 1985). J Ind Microbiol Biotechnol 22(4–5):551–563. doi:10.1038/sj.jim.2900665
Cottrell MT, Kirchman DL (2000) Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl Environ Microbiol 66(4):1692–1697. doi:10.1128/Aem.66.4.1692-1697.2000
Darling JA, Blum MJ (2007) DNA-based methods for monitoring invasive species: a review and prospectus. Biol Invasions 9(7):751–765. doi:10.1007/S10530-006-9079-4
Darling JA, Mahon AR (2011) From molecules to management: adopting DNA-based methods for monitoring biological invasions in aquatic environments. Environ Res 111(7):978–988. doi:10.1016/j.envres.2011.02.001
Decho AW (1990) Microbial exopolymer secretions in ocean environments—their role(s) in food webs and marine processes. Oceanogr Mar Biol 28:73–153
Degnan PH, Ochman H (2012) Illumina-based analysis of microbial community diversity. ISME J 6(1):183–194. doi:10.1038/ismej.2011.74
Dorigo U, Volatier L, Humbert JF (2005) Molecular approaches to the assessment of biodiversity in aquatic microbial communities. Water Res 39(11):2207–2218. doi:10.1016/j.watres.2005.04.007
Doyle TK, Houghton JDR, McDevitt R, Davenport J (2006) Hays GC (2007) The energy density of jellyfish: estimates from bomb-calorimetry and proximate-composition. J Exp Mar Biol Ecol 343(2):239–252. doi:10.1016/J.Jembe.12.010
Duckworth AW, Grant WD, Jones BE, Meijer D, Marquez MC, Ventosa A (2000) Halomonas magadii sp nov., a new member of the genus Halomonas, isolated from a soda lake of the East African Rift Valley. Extremophiles 4(1):53–60
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26(19):2460–2461. doi:10.1093/bioinformatics/btq461
Estoup A, Guillemaud T (2010) Reconstructing routes of invasion using genetic data: why, how and so what? Mol Ecol 19(19):4113–4130. doi:10.1111/J.1365-294x.2010.04773.X
Franzmann PD, Burton HR, McMeekin TA (1987) Halomonas subglaciescola, a new species of halotolerant bacteria isolated from Antarctica. Int J Syst Bacteriol 37(1):27–34. doi:10.1099/00207713-37-1-27
Geller JB, Darling JA, Carlton JT (2010) Genetic perspectives on marine biological invasions. Ann Rev Mar Sci 2:367–393. doi:10.1146/annurev.marine.010908.163745
Ghabooli S, Shiganova TA, Briski E, Piraino S, Fuentes V, Thibault-Botha D, Angel DL, Cristescu ME, MacIsaac HJ (2013) Invasion pathway of the ctenophore Mnemiopsis leidyi in the Mediterranean Sea. PloS ONE 8 (11). doi:10.1371/journal.pone.0081067
Gilbert JA, Meyer F, Jansson J, Gordon J, Pace N, Tiedje J, Ley R, Fierer N, Field D, Kyrpides N, Glockner FO, Klenk HP, Wommack KE, Glass E, Docherty K, Gallery R, Stevens R, Knight R (2010) The Earth Microbiome Project: meeting report of the “1 EMP meeting on sample selection and acquisition” at Argonne National Laboratory October 6 2010. Stand Genomic Sci 3(3):249–253. doi:10.4056/aigs.1443528
Glockner FO, Fuchs BM, Amann R (1999) Bacterioplankton compositions of lakes and oceans: a first comparison based on fluorescence in situ hybridization. Appl Environ Microbiol 65(8):3721–3726
Hajibabaei M, Shokralla S, Zhou X, Singer GA, Baird DJ (2011) Environmental barcoding: a next-generation sequencing approach for biomonitoring applications using river benthos. PLoS ONE 6(4):e17497. doi:10.1371/journal.pone.0017497
Hanfling B (2007) Understanding the establishment success of non-indigenous fishes: lessons from population genetics. J Fish Biol 71:115–135. doi:10.1111/J.1095-8649.2007.01685.X
Hansson LJ, Norrman B (1995) Release of dissolved organic-carbon (DOC) by the scyphozoan jellyfish Aurelia-aurita and its potential influence on the production of planktic bacteria. Mar Biol 121(3):527–532. doi:10.1007/Bf00349462
Holst S, Jarms G (2007) Substrate choice and settlement preferences of planula larvae of five Scyphozoa (Cnidaria) from German Bight. North Sea. Marine Biology 151(3):863–871. doi:10.1007/s00227-006-0530-y
Hosoya S, Arunpairojana V, Suwannachart C, Kanjana-Opas A, Yokota A (2006) Aureispira marina gen. nov., sp nov., a gliding, arachidonic acid-containing bacterium isolated from the southern coastline of Thailand. Int J Syst Evol Microbiol 56:2931–2935. doi:10.1099/Ijs.0.64504-0
Ivanova EP, Sawabe T, Gorshkova NM, Svetashev VI, Mikhailov VV, Nicolau DV, Christen R (2001) Shewanella japonica sp nov. Int J Syst Evol Microbiol 51:1027–1033
Ivanova EP, Nedashkovskaya OI, Zhukova NV, Nicolau DV, Christen R, Mikhailov VV (2003) Shewanella waksmanii sp nov., isolated from a Sipuncula (Phascolosoma japonicum). Int J Syst Evol Microbiol 53:1471–1477. doi:10.1099/ijs.0.02630-0
Ivanova EP, Flavier S, Christen R (2004a) Phylogenetic relationships among marine Alteromonas-like proteobacteria: emended description of the family Alteromonadaceae and proposal of Pseudoalteromonadaceae fam. nov., Colwelliaceae fam. nov., Shewanellaceae fam. nov., Montellaceae fam. nov., Ferrimonadaceae fam. nov., Idiomarinaceae fam. nov and Psychromonadaceae fam. nov. Int J Syst Evol Microbiol 54:1773–1788. doi:10.1099/ijs.0.02997-0
Ivanova EP, Gorshkova NM, Bowman JP, Lysenko AM, Zhukova NV, Sergeev AF, Mikhailov VV, Nicolau DV (2004b) Shewanella pacifica sp nov., a polyunsaturated fatty acid-producing bacterium isolated from sea water. Int J Syst Evol Microbiol 54:1083–1087. doi:10.1099/ijs.0.02993-0
Ivanova EP, Nedashkovskaya OI, Sawabe T, Zhukova NV, Frolova GM, Nicolau DV, Mikhailov VV, Bowman JP (2004c) Shewanella affinis sp nov., isolated from marine invertebrates. Int J Syst Evol Microbiol 54:1089–1093. doi:10.1099/ijs.0.02992-0
Karp A, Edwards KJ, Bruford M, Funk S, Vosman B, Morgante M, Seberg O, Kremer A, Boursot P, Arctander P, Tautz D, Hewitt GM (1997) Molecular technologies for biodiversity evaluation: opportunities and challenges. Nat Biotechnol 15(7):625–628. doi:10.1038/nbt0797-625
Kemp PF, Aller JY (2004) Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us. FEMS Microbiol Ecol 47(2):161–177. doi:10.1016/S0168-6496(03)00257-5
Kim JJ, Kim JH, Kwon YK, Kwon KK, Yang SH, Jang J, Heo SJ, Park HS, Jung WK, Lee Y, Kang DH, Oh C (2013) Algivirga pacifica gen. nov., sp. nov., a novel agar-degrading marine bacterium of the family Flammeovirgaceae isolated from Micronesia. Curr Microbiol 67(6):742–747. doi:10.1007/s00284-013-0429-z
Kirchman DL (2002) The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol Ecol 39(2):91–100. doi:10.1111/j.1574-6941.2002.tb00910.x
Kisand V, Valente A, Lahm A, Tanet G, Lettieri T (2012) Phylogenetic and functional metagenomic profiling for assessing microbial biodiversity in environmental monitoring. PLoS ONE 7(8):e43630. doi:10.1371/journal.pone.0043630
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glockner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41(1):e1. doi:10.1093/nar/gks808
Kowalchuk GA, Jones SE, Blackall LL (2008) Microbes orchestrate life on Earth. ISME J 2(8):795–796. doi:10.1038/ismej.2008.61
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79(17):5112–5120. doi:10.1128/AEM.01043-13
LaDouceur EEB, Garner MM, Wynne J, Fish S, Adams L (2013) Ulcerative Umbrellar Lesions in Captive Moon Jelly (Aurelia aurita) Medusae. Vet Pathol 50(3):434–442. doi:10.1177/0300985812461363
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–359. doi:10.1038/nmeth.1923
Lau SC, Tsoi MM, Li X, Plakhotnikova I, Dobretsov S, Wu M, Wong PK, Pawlik JR, Qian PY (2006) Description of Fabibacter halotolerans gen. nov., sp. nov. and Roseivirga spongicola sp. nov., and reclassification of [Marinicola] seohaensis as Roseivirga seohaensis comb. nov. Int J Syst Evol Microbiol 56 (Pt 5):1059–1065. doi:10.1099/ijs.0.64104-0
Ley RE, Harris JK, Wilcox J, Spear JR, Miller SR, Bebout BM, Maresca JA, Bryant DA, Sogin ML, Pace NR (2006) Unexpected diversity and complexity of the Guerrero Negro hypersaline microbial mat. Appl Environ Microbiol 72(5):3685–3695. doi:10.1128/Aem.72.5.3685-3695.2006
Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M (2012) Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012:251364. doi:10.1155/2012/251364
Lo WT, Purcell JE, Hung JJ, Su HM, Hsu PK (2008) Enhancement of jellyfish (Aurelia aurita) populations by extensive aquaculture rafts in a coastal lagoon in Taiwan. ICES J Mar Sci 65(3):453–461
Loman NJ, Constantinidou C, Chan JZ, Halachev M, Sergeant M, Penn CW, Robinson ER, Pallen MJ (2012) High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol 10(9):599–606. doi:10.1038/nrmicro2850
Luo C, Tsementzi D, Kyrpides N, Read T, Konstantinidis KT (2012) Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 7(2):e30087. doi:10.1371/journal.pone.0030087
Magoc T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27(21):2957–2963. doi:10.1093/bioinformatics/btr507
Malej A, Turk V, Lucic D, Benovic A (2007) Direct and indirect trophic interactions of Aurelia sp (Scyphozoa) in a stratified marine environment (Mljet Lakes, Adriatic Sea). Mar Biol 151(3):827–841. doi:10.1007/s00227-006-0503-1
Malej A, Kogovsek T, Ramsak A, Catenacci L (2012) Blooms and population dynamics of moon jellyfish in the northern Adriatic. Cah Biol Mar 53(3):337–342
Manini EBP, D’Adamo R, Spagnoli F, Danovaro. R. (2005) Lagoon of Varano. In: Giordani G, Viaroli P, Swaney DP, Murray CN, Zaldıvar JM, Marshall JI Crossland (Eds.), LOICZ Reports & Studies No. 28, LOICZ, Texel, The Netherlands 55–58
Melaku Canu D, Solidoro C, Umgiesser G, Cucco A, Ferrarin C (2012) Assessing confinement in coastal lagoons. Mar Pollut Bull 64(11):2391–2398
Mellado E, Moore ERB, Nieto JJ, Ventosa A (1995) Phylogenetic Inferences and Taxonomic Consequences of 16S Ribosomal DNA Sequence Comparison of Chromohalobacter marismortui, Volcaniella eurihalina, and Deleya salina and Reclassification of V. eurihalina as Halomonas eurihalina comb. nov. Int J Syst Bacteriol 45(4):712–716. doi:10.1099/00207713-45-4-712
Mende DR, Sunagawa S, Zeller G, Bork P (2013) Accurate and universal delineation of prokaryotic species. Nat Methods 10(9):881–884. doi:10.1038/nmeth.2575
Milisenda G, Rosa S, Fuentes VL, Boero F, Guglielmo L, Purcell JE, Piraino S (2014) Jellyfish as Prey: frequency of Predation and Selective Foraging of Boops boops (Vertebrata, Actinopterygii) on the Mauve Stinger Pelagia noctiluca (Cnidaria, Scyphozoa). PLoS ONE 9(4):e94600. doi:10.1371/journal.pone.0094600
Mills CE (2001) Jellyfish blooms: are populations increasing globally in response to changing ocean conditions? Hydrobiologia 451(1–3):55–68. doi:10.1023/a:1011888006302
Miura O (2007) Molecular genetic approaches to elucidate the ecological and evolutionary issues associated with biological invasions. Ecol Res 22(6):876–883. doi:10.1007/S11284-007-0389-5
Mormile MR, Romine MF, Garcia T, Ventosa A, Bailey TJ, Peyton BM (1999) Halomonas campisalis sp nov., a denitrifying, moderately haloalkaliphilic bacterium. Syst Appl Microbiol 22(4):551–558
Muirhead JR, Gray DK, Kelly DW, Ellis SM, Heath DD, Macisaac HJ (2008) Identifying the source of species invasions: sampling intensity vs. genetic diversity. Mol Ecol 17(4):1020–1035. doi:10.1111/J.1365-294x.2008.03669.X
Ogawa H, Amagai Y, Koike I, Kaiser K, Benner R (2001) Production of refractory dissolved organic matter by bacteria. Science 292(5518):917–920. doi:10.1126/science.1057627
Piraino S, Aglieri G, Martell L, Mazzoldi C, Melli V, Milisenda G, Scorrano S, Boero F (2014) Pelagia benovici sp nov (Cnidaria, Scyphozoa): a new jellyfish in the Mediterranean Sea. Zootaxa 3794(3):455–468
Pitt KA, Welsh DT, Condon RH (2009) Influence of jellyfish blooms on carbon, nitrogen and phosphorus cycling and plankton production. Hydrobiologia 616:133–149. doi:10.1007/s10750-008-9584-9
Pommier T, Pinhassi J, Hagstrom A (2005) Biogeographic analysis of ribosomal RNA clusters from marine bacterioplankton. Aquat Microb Ecol 41(1):79–89. doi:10.3354/ame041079
Purcell JE (2012) Jellyfish and ctenophore blooms coincide with human proliferations and environmental perturbations. Annu Rev Mar Sci 4:209–235
Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y (2012) A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13:341. doi:10.1186/1471-2164-13-341
Quesada E, Ventosa A, Ruiz-Berraquero F, Ramos-Cormenzana A (1984) Deleya halophila, a new species of moderately halophilic bacteria. Int J Syst Bacteriol 34(3):287–292. doi:10.1099/00207713-34-3-287
Quesada E, Valderrama MJ, Bejar V, Ventosa A, Gutierrez MC, Ruiz-Berraquero F, Ramos-Cormenzana A (1990) Volcaniella eurihalina gen. nov., sp. nov., a moderately halophilic nonmotile Gram-negative rod. Int J Syst Bacteriol 40(3):261–267. doi:10.1099/00207713-40-3-261
Rakow KC, Graham WM (2006) Orientation and swimming mechanics by the scyphomedusa Aurelia sp in shear flow. Limnol Oceanogr 51(2):1097–1106
Reichenbach H (1991) The order Cytophagales. In: Balows A, Tru¨per HG, Dworkin M, Harder W, Schleifer K-H (eds) The prokaryotes, 2nd edn. Springer Verlag, New York, pp 3631–3675
Riemann L, Steward GF, Azam F (2000) Dynamics of bacterial community composition and activity during a mesocosm diatom bloom. Appl Environ Microbiol 66(2):578–587. doi:10.1128/Aem.66.2.578-587.2000
Riemann L, Titelman J, Bamstedt U (2006) Links between jellyfish and microbes in a jellyfish dominated fjord. Mar Ecol Prog Ser 325:29–42. doi:10.3354/Meps325029
Rubin BE, Gibbons SM, Kennedy S, Hampton-Marcell J, Owens S, Gilbert JA (2013) Investigating the impact of storage conditions on microbial community composition in soil samples. PLoS ONE 8(7):e70460. doi:10.1371/journal.pone.0070460
Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, Wu D, Eisen JA, Hoffman JM, Remington K, Beeson K, Tran B, Smith H, Baden-Tillson H, Stewart C, Thorpe J, Freeman J, Andrews-Pfannkoch C, Venter JE, Li K, Kravitz S, Heidelberg JF, Utterback T, Rogers YH, Falcon LI, Souza V, Bonilla-Rosso G, Eguiarte LE, Karl DM, Sathyendranath S, Platt T, Bermingham E, Gallardo V, Tamayo-Castillo G, Ferrari MR, Strausberg RL, Nealson K, Friedman R, Frazier M, Venter JC (2007) The Sorcerer II global ocean sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 5(3):e77. doi:10.1371/journal.pbio.0050077
Schauer M, Hahn MW (2005) Diversity and phylogenetic affiliations of morphologically conspicuous large filamentous bacteria occurring in the pelagic zones of a broad spectrum of freshwater habitats. Appl Environ Microbiol 71(4):1931–1940. doi:10.1128/Aem.71.4.1931-1940.2005
Schuett C, Doepke H (2010) Endobiotic bacteria and their pathogenic potential in cnidarian tentacles. Helgoland Mar Res 64(3):205–212. doi:10.1007/S10152-009-0179-2
Scorrano S (2014) Impacts of Aurelia sp. 1 outbreaks in Mediterranean coastal lagoon (Varano, SE Adriatic Coast). Ph.D. Thesis in ecology and management of biological resources, Università degli Studi della Tuscia, Viterbo, Italy
Sharma KK, Kalawat U (2010) Emerging infections: shewanella—a series of five cases. Journal of laboratory physicians 2(2):61–65. doi:10.4103/0974-2727.72150
Sieburth J (1979) Sea microbes. Oxford University Press, New York, p 491
Simidu U, Kita-Tsukamoto K, Yasumoto T, Yotsu M (1990) Taxonomy of four marine bacterial strains that produce tetrodotoxin. Int J Syst Bacteriol 40(4):331–336. doi:10.1099/00207713-40-4-331
Skerratt JH, Bowman JP, Nichols PD (2002) Shewanella olleyana sp nov., a marine species isolated from a temperate estuary which produces high levels of polyunsaturated fatty acids. Int J Syst Evol Microbiol 52:2101–2106. doi:10.1099/ijs.0.02351-0
Spagnoli F, Specchiulli A, Scirocco T, Carapella G, Villani P, Casolino G, Schiavone P, Franchi M (2002) The Lago di Varano: hydrologic characteristics and sediment composition. Mar Ecol P.S.Z.N I 23:384–394. doi:10.1111/J.1439-0485.2002.Tb00036.X
Specchiulli A, Focardi S, Renzi M, Scirocco T, Cilenti L, Breber P, Bastianoni S (2008) Environmental heterogeneity patterns and assessment of trophic levels in two Mediterranean lagoons: Orbetello and Varano, Italy. Sci Total Environ 402(2–3):285–298. doi:10.1016/J.Scitotenv.04.052
Stabili L, Cavallo RA (2011) Microbial pollution indicators and culturable heterotrophic bacteria in a Mediterranean area (Southern Adriatic Sea Italian coasts). J Sea Res 65(4):461–469. doi:10.1016/j.seares.2011.04.010
Stabili L, Caprioli R, Kos Kramar M, Scorrano S, Boero F, Turk V, Piraino S (2013) Jellyfish and the bacterial community of Varano lagoon. Paper presented at the First EMBO Conference on Aquatic Microbial Ecology SAME13, p 202 (abstract)
Stecher B, Chaffron S, Kappeli R, Hapfelmeier S, Freedrich S, Weber TC, Kirundi J, Suar M, McCoy KD, von Mering C, Macpherson AJ, Hardt WD (2010) Like will to like: abundances of closely related species can predict susceptibility to intestinal colonization by pathogenic and commensal bacteria. PLoS Pathog 6(1):e1000711. doi:10.1371/journal.ppat.1000711
Stierle AC, Cardellina JH II, Singleton FL (1988) A marine Micrococcus produces metabolites ascribed to the sponge Tedania ignis. Experientia 44(11–12):1021
Thomas F, Hehemann JH, Rebuffet E, Czjzek M, Michel G (2011) Environmental and gut bacteroidetes: the food connection. Front Microbiol 2:93. doi:10.3389/fmicb.2011.00093
Thomas T, Gilbert J, Meyer F (2012) Metagenomics—a guide from sampling to data analysis. Microb Inform Exp 2(1):3. doi:10.1186/2042-5783-2-3
Tinta T, Malej A, Kos M, Turk V (2010) Degradation of the Adriatic medusa Aurelia sp by ambient bacteria. Hydrobiologia 645(1):179–191. doi:10.1007/s10750-010-0223-x
Tinta T, Kogovsek T, Malej A, Turk V (2012) Jellyfish modulate bacterial dynamic and community structure. PloS ONE 7 (6). doi:10.1371/journal.pone.0039274
Titelman J, Riemann L, Sornes TA, Nilsen T, Griekspoor P, Bamstedt U (2006) Turnover of dead jellyfish: stimulation and retardation of microbial activity. Mar Ecol Prog Ser 325:43–58. doi:10.3354/Meps325043
Valderrama MJ, Quesada E, Bejar V, Ventosa A, Gutierrez MC, Ruiz-Berraquero F, Ramos-Cormenzana A (1991) Deleya salina sp. nov., a moderately halophilic Gram-negative bacterium. Int J Syst Bacteriol 41(3):377–384. doi:10.1099/00207713-41-3-377
van Veen WL, van der Kooij D, Geuze EC, van der Vlies AW (1973) Investigations on the sheathed bacterium Haliscomenobacter hydrossis gen.n., sp.n., isolated from activated sludge. Antonie Van Leeuwenhoek 39(2):207–216
Venkateswaran K, Shimada A, Maruyama A, Higashihara T, Sakou H, Maruyama T (1993) Microbial characteristics of palau jellyfish lake. Can J Microbiol 39(5):506–512
Venkateswaran K, Dollhopf ME, Aller R, Stackebrandt E, Nealson KH (1998) Shewanella amazonensis sp. nov., a novel metal-reducing facultative anaerobe from Amazonian shelf muds. Int J Syst Bacteriol 48:965–972
Vreeland RH, Litchfield CD, Martin EL, Elliot E (1980) Halomonas elongata, a new genus and species of extremely salt-tolerant bacteria. Int J Syst Bacteriol 30(2):485–495. doi:10.1099/00207713-30-2-485
Yamamoto J, Hirose M, Ohtani T, Sugimoto K, Hirase K, Shimamoto N, Shimura T, Honda N, Fujimori Y, Mukai T (2008) Transportation of organic matter to the sea floor by carrion falls of the giant jellyfish Nemopilema nomurai in the Sea of Japan. Mar Biol 153(3):311–317. doi:10.1007/s00227-007-0807-9
Yoon JH, Kang KH, Oh TK, Park YH (2004a) Shewanella gaetbuli sp nov., a slight halophile isolated from a tidal flat in Korea. Int J Syst Evol Microbiol 54:487–491. doi:10.1099/ijs.0.02731-0
Yoon JH, Yeo SH, Kim IG, Oh TK (2004b) Shewanella marisflavi sp nov and Shewanella aquimarina sp nov., slightly halophilic organisms isolated from sea water of the Yellow Sea in Korea. Int J Syst Evol Microbiol 54:2347–2352. doi:10.1099/ijs.0.63198-0
Yoon J, Katsuta A, Kasai H (2012) Rubidimonas crustatorum gen. nov., sp nov., a novel member of the family Saprospiraceae isolated from a marine crustacean. Antonie Von Leeuwenhoek 101(3):461–467
Zehr JP (2010) Microbes in Earth’s Aqueous Environments. Frontiers in Microbiology 1:4. doi:10.3389/fmicb.2010.00004
Zinger L, Gobet A, Pommier T (2012) Two decades of describing the unseen majority of aquatic microbial diversity. Mol Ecol 21(8):1878–1896. doi:10.1111/j.1365-294X.2011.05362.x
Acknowledgments
The publication of this paper is supported by CONISMA, the Italian National Interuniversity Consortium for Marine Sciences, which received funding from the European Community’s Seventh Framework Programme (FP7/2007–2013) for the project VECTORS (http://www.marine-vectors.eu). This paper stems from the International workshop MOLTOOLS (Molecular Tools for Monitoring Marine Invasive Species), held in Lecce, Italy, in September 2012. Logistic/technical support was also provided by the FP7 EU projects COCONET, PERSEUS, the ENPI CBCMED programme MEDJELLYRISK, and by the Italian Flagship project RITMARE. The authors thank Jennifer Purcell for critical reading and English revision of the manuscript. The experimental activity was carried out in the Molecular Biodiversity Lab of ESFRI Infrastructure Lifewatch (Bari, Italy).
Author information
Authors and Affiliations
Corresponding author
Additional information
Caterina Manzari, Bruno Fosso and Marinella Marzano have contributed equally to this work.
Stefano Piraino and Graziano Pesole are senior authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

{kind=link}
{kind=link}
Cite this article
Manzari, C., Fosso, B., Marzano, M. et al. The influence of invasive jellyfish blooms on the aquatic microbiome in a coastal lagoon (Varano, SE Italy) detected by an Illumina-based deep sequencing strategy. Biol Invasions 17, 923–940 (2015). https://doi.org/10.1007/s10530-014-0810-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-014-0810-2