
Abstract
Native to Southeast Asia, the spotted wing drosophila (SWD), Drosophila suzukii Matsumura, rapidly invaded America and Europe in the past 20 years. As a crop pest of soft-skinned fruits with a wide range of host plants, it threatens the fruit industry worldwide, causing enormous economic losses. To control this invasive pest species, an understanding of its population dynamics and structure is necessary. Here, we report the population genetics and development of SWD in Germany from 2017–19 using microsatellite markers over 11 different sample sites. It is the first study that examines SWD’s genetic changes over 3 years compared to multiple international SWD laboratory strains. Results show that SWD populations in Germany are highly homogenous without differences between populations or years, which indicates that populations are well adapted, migrate freely, and multiple invasions from outside Germany either did not take place or are negligible. Such high genetic variability and migration between populations could allow for a fast establishment of the pest species. This is especially problematic with regard to the ongoing spread of this invasive species and could bear a potential for developing pesticide resistance, which could increase the impact of the SWD further in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Key message
-
Drosophila suzukii (Spotted Wing Drosophila) is established and migrates freely in Germany.
-
Homogenous populations across Germany were found over the 3-year sampling period.
-
Multiple reinvasions of Drosophila suzukii either do not take place or are negligible.
Introduction
The spotted wing drosophila (SWD), Drosophila suzukii Matsumura, became a severe invasive pest species in America and Europe. First descriptions of D. suzukii in Japan date back to 1931, recorded by Matsumura (Matsumura 1931). At this time, SWD was present throughout Japan, Korea, and China (Hauser 2011). Through its rapid spread across North America, Africa and Europe in the last few years, SWD has drawn much attention as a crop pest in affected countries (Hauser 2011; Calabria et al. 2012; Asplen et al. 2015; Boughdad et al. 2021). SWD spread all over the continent and belongs to the commonly found drosophilids in South and Central Europe (Cini et al. 2012; Calabria et al. 2012). The first specimens of SWD from Germany were caught in 2011 in Rhineland-Palatinate, Baden-Württemberg, and at Lake Constance (Bavaria and Baden-Württemberg) (Vogt et al. 2012).
In contrast to the vinegar fly Drosophila melanogaster, SWD is a crop pest for soft-skinned fruits like cherries, blueberries, blackberries, grapes, and strawberries. Female flies possess a serrated ovipositor with which they pierce the skin of ripening fruits. Hatching larvae feed on fruit pulp and make the fruits unmarketable, causing substantial economic losses (Bolda et al. 2010; Mazzi et al. 2017). Bolda et al. (2010) calculated that the revenue losses in raspberries and blackberries in California totaled approximately $63.2 million in 2008 alone. Besides the larval damage, probing and rupturing of the fruit skin by the female ovipositor can entail several other problems like fungal infections that cause additional damage to the fruits (Rombaut et al. 2017; Ioriatti et al. 2018). An essential part of tackling an invasive pest like SWD is understanding its biology and answering, e.g., questions on the threat, SWD poses for individual fruit crops (Lee et al. 2011; Bellamy et al. 2013). Therefore, monitoring of fly populations is continuously conducted in Germany, and institutions provide growers with information on best practice control methods (Briem et al. 2015, 2018). Many approaches to develop SWD control have focused on live traps and pesticide applications so far. In contrast, other practices such as mass trapping and biological control methods using imported or native parasitoids could be central for future pest management strategies (Gabarra et al. 2015; Rossi Stacconi et al. 2015).
Efforts of pest control applications can be complemented and improved by understanding genetic development. Knowledge about invasion history and the genetic differences between populations can identify introduction pathways, improve testing, and help integrated pest management strategies in ultimately avoiding multiple (re)introductions (Estoup and Guillemaud 2010). Stabelli et al. (2020) reported that SWD populations from separate geographic areas exhibit different genotypic and phenotypic traits pointing at the importance of understanding intraspecific variability in pest species for pest control. The reconstruction of invasion pathways is particularly crucial to uncover and understand potential patterns in the spread of an invasive species. It is thought that pest organisms are distributed beyond their native ranges through trade by contaminations of traded goods or as stowaways (Chapman et al. 2017). Information on transport and introduction of non-native species is of great importance, because the management of invasive pest species is most effective during the beginning of an invasion, if, e.g., biosecurity measures would be adopted at transport routes (Rout et al. 2011; Chapman et al. 2016). In a recent study, it was shown that the latest invasion of SWD in Argentina could be ascribed to fruit trade from freshly invaded areas in North and South America and not from its native range in Asia (de la Vega et al. 2020). Data from Ukraine also implies that SWD invasion in Europe can be ascribed to multiple sources, along with possible recurrent introductions (Lavrinienko et al. 2017). The results from these two studies show how the identification of invasion routes is an important issue, especially from a pest management perspective, and they point out the problematic link between global trade and pest invasiveness. The reconstruction of invasion routes for international trade can be challenging, though, considering the sheer amount of fruit transports through global trading. For example, Germany imported 51,776 tons of fresh fruits in the year 2017 alone (FAO 2021). Studies on population genetics have proven beneficial for biological control methods like the sterile insect technique (SIT) (Lanzavecchia et al. 2014). SIT is a biological method for pest control in which sterilized male insects are released in a field population to reduce reproduction by infertile mating. Since gene flow can vary between natural populations, locally adapted and isolated populations can occur. In these populations, SIT can be impaired by mating barriers, making them less effective. Population genetics can be used to trace changes in strain efficacy, which is an assertion of how well a strain would perform in the field. It can also help to improve the competitiveness of laboratory strains that are sterilized and released and can be used in monitoring programs to differentiate released laboratory insects from wild ones (Aketarawong et al. 2011, 2014; Zygouridis et al. 2014; Azrag et al. 2016).
One way to characterize genetic relationships between populations is by using microsatellite or simple sequence repeat (SSR) markers. Microsatellites are repetitive DNA sequences that have high mutation rates, which can lead to high polymorphism within populations and a rapid genetic differentiation between distinct populations (Schlötterer 2000; Selkoe and Toonen 2006). Variations in the number of repetitions generate different alleles. This long-established method in population genetics is used regularly due to its cost-effectiveness and informative content (Jarne and Lagoda 1996; Schlötterer 2004). For D. suzukii, a set of 28 microsatellite markers was previously developed and used to characterize genetic variation within Hawaiian and French populations (Fraimout et al. 2015). A subset of those markers was then utilized to get an insight into genetic variability in Italian populations of D. suzukii (Tait et al. 2017). Those populations showed extensive genetic similarity, except for a population from Sicily, confirming isolation relative to the mainland. Whereas the technique has been widely used to describe population variation and genetic differences in SWD, only few research has investigated differences that occur over consecutive years when this pest species begins to become established. Bahder et al. 2015 evaluated the genetic variation in SWD populations from California and Washington collected across three years and showed that the population from Washington had undergone a significant bottleneck compared to the coastal California population. These studies show that the use of microsatellite markers for population structure and genetic diversity analysis is an important part of understanding the biology of an invasive species. Because little is known about the genetic diversity of SWD populations in Germany, the goal of this study was to determine intrapopulation and interpopulation genetic diversity between different populations across the whole country. Moreover, it was of interest, if diverse genetically defined populations exist and if they exhibit a geographical pattern. On the one hand, we expected some differences between northern and southern locations, since the North and South of Germany are two distinct major geographical regions with different geographic aspects and different climates with a more maritime climate in the North and an increasingly subcontinental climate toward the South. On the other hand, based on the results of Tait et al. (2017) from Italy, we expected low differences between sample sites, since the distances between locations are relatively low. An additional comparison of data over several years can provide information about population development and dynamics. In respect to pest control, it would be beneficial to know, if an annual reinvasion from other European countries or warmer regions in Germany takes place or if SWD overwinters locally and reemerges in spring. It is known that SWD overwinter as adults and can recover quickly (Dalton et al. 2011; Hamby et al. 2014; Stephens et al. 2015). Thus, we expected that a reemergence from locally overwintering individuals is more likely and, if differences between years occur, that they turn out to be small. Invasive species often show a high genetic divergence between their invasive and native populations, which allows them to adapt faster to new environments (Guo et al. 2018; Xia et al. 2020). High genetic diversity in Germany would implicate a well-established population, while reduced genetic diversity, for example through a bottleneck effect, in contrast, would correspond to reduced adaptability or at least to a recent invasion at that specific locality (Schrader et al. 2014). Therefore, we investigated D. suzukii populations for three years using 14 microsatellite markers designed by Fraimout et al. (2015). This is the first study to provide insights into population genetics of D. suzukii on a large-scale level in Germany as well as a comparison over a multi-year period. Besides, different laboratory strains from Europe and America were included in the comparison and a new laboratory strain that derived from one of the German field populations was established to illustrate the effect of laboratory breeding and change of genetic markers over time. As a result, this strain functioned as a link between field collections and the laboratory strains.
Material and methods
Drosophila suzukii collection and identification
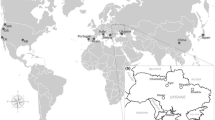
Adult D. suzukii from 11 locations in Germany were collected between June and September in 2017, 2018, and 2019 by collecting fruit samples infested with D. suzukii eggs, larvae, or pupae. Location sites were mapped using geographic coordinates with Simplemappr (Shorthouse 2010), assuring that each year fruits from the same locations were collected (Fig. 1). If possible, different kind of fruits from various shrubs or trees were sampled within a radius of approximately 20 km at each location, to prevent sampling of related individuals. Blackberries showed the highest amount of infestation followed by raspberries and cherries. Elderberries were collected only in a few cases since the degree of infestation was lower than in cherries. Strawberries were not infested. The number of sampled fruits varied between years and locations (Table 1). The German samples' names result from the vehicle registration plate of the respective district and the sampling year. Fruits were kept in the laboratory until adult flies emerged. Adult D. suzukii were identified according to Hauser (2011) and documented using a Keyence VHX-5000 (Keyence Corporation, Osaka, Japan). No samples were available from FF in 2017, HB in 2018 and 2019, and HH in 2017 and 2018 (Fig. 1, Table 1). From each geographical population and year, 20 individuals were tested, except for HB17, with only ten individuals available.

Sample sites and sampling time points of D. suzukii wild populations in Germany. Population location acronym, location, coordinates and sampling year and month are listed in the table (left). The map (right side) was generated by SimpleMappr and coordinates listed in the table
In addition, 160 specimens from eight laboratory strains (LS_USA, LS_Canada, LS_Italy, LS_Frankfurt, LS_France, LS_Valsugana, LS_HG18, and LS_HG19) were used as outgroups. LS_USA was established in 2010 from a field collection in North Carolina (Stockton et al. 2020), LS_Canada was started in 2012 (Jakobs et al. 2015; Renkema et al. 2015), LS_Italy was kept in the laboratory since 2014 and LS_Frankfurt was established in 2016 (Lee and Vilcinskas 2017). LS_France was kindly provided by Eric Marois and originated from Strasbourg (France). Alberto Grassi provided LS_Valsugana from Valsugana in Italy. Both were collected in 2018 and kept in the laboratory since. The laboratory strains LS_HG18 and LS_HG19 originated from flies collected from the location Bad Homburg (HG) in 2017 that were sampled for analysis after one (LS_HG18) and two years (LS_HG19) in culture, respectively. D. suzukii laboratory strains were maintained on standard Drosophila medium at 25 °C and 55% humidity with a 12 h-photoperiod and transferred to fresh media every week. A sample size estimation was conducted to determine the minimum number of observations required for our experiment. Under the assumption, that the population standard deviation of allele size is at most seven (based on preliminary exploratory data analyses) and under the requirement that a 90%-confidence interval (CI) for the population mean of allele size has a length of at most 10 with a probability of 80% (“precision power”), a sample size of at least 19 was necessary. Based on this sample size estimation, we chose to use 20 individuals from each location and year with the exception of HB17, where only 10 individuals were available. The final data set included 550 individuals from Germany and 160 individuals from laboratory strains (Table 1). German populations and laboratory outgroups were analyzed using 14 microsatellite markers.
DNA extraction and microsatellite analysis
Total genomic DNA was extracted from 20 or 10 individuals of each geographical population and laboratory strain in single reactions (Table 1). Samples were placed in lysis tubes with 1.4 mm ceramic spheres (Lysing Matrix D bulk, MP Biomedicals, Solon, OH), 200 µl sterile filtered homogenization buffer added (1 M Tris–HCl (pH 7.5), 5 M NaCl, 0.5 M EDTA (pH 8), 0.3 M spermine tetra-HCl, 1 M spermidine tri-HCl, 1 g sucrose), and then homogenized at 6000 rpm for 40 s in a Fast Prep-24™ (MP Biomedicals, Solon, OH). Afterward, 200 µl lysis buffer (1 M Tris–HCl (pH 9.0), 0.5 M EDTA (pH 8.0), 10% SDS, 1 g sucrose) was added and incubated at 70 °C for 10 min. 60 µl of 8 M KOAc was added, and tubes were stored on ice for 30 min. After transferring the suspension to a new tube, samples were centrifuged at 12,000 rpm for 10 min at 4 °C. DNA was precipitated with two volumes of ice-cold 100% EtOH and stored at -20 °C overnight. DNA was pelleted by centrifugation at 12,000 rpm for 40 min at 4 °C. The pellet was washed with 30 µl of 70% ice-cold ethanol for 10 min while centrifuging at 12,000 rpm at 4 °C. Pellets were air-dried and resuspended in 50 µl H2O. DNA concentration was measured using an Epoch Microplate Spectrophotometer (BioTek, Winooski, VT, USA). Samples were stored at -20 °C.
SSR markers were previously characterized (Fraimout et al. 2015). Of the 28 published SSRs, 17 dinucleotide markers were selected for initial testing with genomic DNA from LS_USA. Each of the published primer pairs was tested beforehand in single PCR reactions using the Platinum Taq DNA polymerase (Thermo Fisher Scientific, MA, USA) following the manufacturer’s protocol with a primer annealing temperature of 57 °C. PCR products were purified with Zymo Clean and Concentrator-5 spin columns (Zymo Research, Orange, CA). Purified PCR products were cloned into the pCR4-TOPO TA vector (Thermo Fisher Scientific, MA, USA) using standard protocols. The sequencing reaction was carried out by Macrogen (Amsterdam, Netherlands) and analyzed with Geneious Prime 2019.2 software. Three of 17 tested loci were not included in further experiments due to amplification problems and null alleles, leaving 14 markers for analysis (Online Resource Table S1).
DNA amplification for fragment analysis on the population samples was then performed using the Qiagen Multiplex PCR Kit (Qiagen, Hilden, Germany) in 10 μL final reaction volume, containing 1X QIAGEN Multiplex PCR Master Mix, 0.2 μM primer mix, 0.5X Q-Solution, and 100 ng of genomic DNA. The PCR cycling protocol was: 95 °C, 5 min; 32 cycles of 95 °C for 30 s, 57 °C for 90 s, 72 °C for 3 min; final elongation at 72 °C for 30 min, the latter is advised to be used for analysis on capillary sequencers. PCR products were purified using the Zymo Clean and Concentrator-5 Kit (Zymo Research, Orange, CA). Concentration was assessed using electrophoresis on 3% agarose gels, stained with SYBR Safe, and visualized under UV light. Samples were sent for fragment analysis on an ABI 3730 Genetic Analyzer to StarSEQ (Mainz, Germany). GeneScan™-500LIZ™ was used as an internal size standard. Fragments were sized with the Geneious Prime 2019.2 software. If no sample amplification was obtained after three attempts, the locus was classified as missing data.
Statistics on genetic diversity
Population genetic indexes like the number of alleles (Na), the effective number of alleles (Ne), the inbreeding coefficient (FIS) and the FST index (where I stands for individuals, S for subpopulations and T for the total population), number of private alleles (AP), observed and expected heterozygosity and deviations from Hardy–Weinberg-Equilibrium were calculated with GenAlex software v.6.41 (Peakall and Smouse 2012). Polymorphism information content (PIC) was calculated using the Cervus 3.0 software (Kalinowski et al. 2007). Measures of diversity were analyzed using repeated-measures analysis of variance (ANOVA) to determine differences among populations and years. Pairwise comparisons were obtained from posthoc Bonferroni correction. All standard statistical tests were carried out using SigmaPlot 14 software (Systat Software, Inc.).
Statistics—genetic structure
In general, the genetic structure refers to patterns of genetic diversity across multiple populations and subpopulations and provides information on distribution, mating behavior, and potential species and population borders. The genetic structure of D. suzukii populations in Germany was analyzed using a Bayesian approach, a population tree based on allele frequency data, analysis of molecular variance (AMOVA), and Principal coordinate analysis (PCoA). Structure 2.3.4 (Pritchard et al. 2000; Falush et al. 2003) was used to investigate the number of genetically distinct clusters (K) in a data set. Each analysis was run with 1,000,000 Markov Chain Monte Carlo (MCMC) repetitions and a burn-in period of 100,000 repetitions using 20 iterations of K = 1–20. The analysis was run using the admixture ancestry model, based on correlated allele frequencies. Structure Harvester (Web v0.6.94 July 2014) (Earl and von Holdt 2012) was used to detect the most likely K value according to Evanno and Pritchard, assessed through analysis of ΔK, the Dirichlet parameter alpha (α) and LnP(D) distribution plots (Evanno et al. 2005; Hubisz et al. 2009). Pophelper (Francis 2017) was used to align assignment clusters across replicate runs and visualize the results.
An unrooted neighbor-joining (NJ) dendrogram was constructed with PoptreeW (Takezaki et al. 2014) based on Da distance (Nei et al. 1983), which is a genetic dissimilarity coefficient that is based on mutation and drift. It is defined as \({D}_{A}=1-\frac{1}{r}{\sum }_{j}^{r}{\sum }_{j}^{{m}_{j}}\sqrt{{x}_{ij}{y}_{ij}}\), where r is the number of loci used, mj is the number of alleles at the j-th locus, and xij and yij are the frequencies of the i-th allele at the j-th locus in populations X and Y (Nei et al. 1983; Takezaki et al. 2014). A test for robustness was carried out by using a bootstrap value of 10,000. An analysis of molecular variance (AMOVA) was performed using Arlequin v.3.5.2.2 (Excoffier and Lischer 2010). PCoA implemented in GenAIex software v.6.41 (Peakall and Smouse 2012) based on Nei’s genetic distance was used to visualize the genetic relationship between populations.
Geneclass2 (Piry et al. 2004) was run to determine the probability of each individual to originate from the sample area or another reference population. The standard Bayesian criterion of Rannala and Mountain (1997) and the Monte Carlo resampling method of Paetkau et al. (2004) were used with an alpha value of 0.05. Results were based on 10,000 simulated genotypes for each population and a threshold probability value of 0.05. Tables with migration rate values are shown in Online Resource S8.
Bottleneck v.1.2.2 (Piry and Luikart 1999) was used to determine if recent demographic events like expansion or mitigation in population size took place. The two-phase model (TPM) and the stricter stepwise mutation model (SMM) were used with model options for the TPM of 80% single-step mutations, a variance among multiple steps of 12 and with 5,000 iterations. Wilcoxon’s signed-rank test was used to assess the probability of heterozygosity.
Results
Polymorphisms and genetic variability at 14 selected microsatellite loci in Drosophila suzukii
In the first step, we evaluated the genetic variability and the information content of the 14 microsatellite markers used in this study. In total, 115, 107, and 112 alleles were identified in 2017, 2018, and 2019, respectively, and all three years shared 81.82% of the identified alleles. The detailed results for variability indices in the 14 SSR markers are shown in Online Resource S2. The analyzed loci showed a consistently high level of genetic variability throughout the years and populations. Deviation from HWE was tested for all year-locus combinations. A significant difference (p < 0.05) from HWE was observed in 24 of 42 year-locus combinations (Online Resource S3). A reason for the disequilibrium could be the presence of null alleles. Per definition, a microsatellite null allele is an allele at a microsatellite locus that does not amplify to detectable levels in a PCR test. The result is an excess of homozygotes or to be precise a deficiency of observed heterozygotes in a population. Other reasons for a deviation from HWE can be migration, selection, phenotypic assortative mating, inbreeding, genetic drift and small population size.
Polymorphism information content (PIC) was obtained as index for gene abundance. The level of diversity reflects genetic variation in loci (PIC > 0.5 = high polymorphism, 0.5 > PIC > 0.25 = moderate polymorphism, PIC < 0.25 = low polymorphism). PIC across loci ranged from 0.474 (DS11) to 0.836 (DS07). All loci except for DS11 showed high polymorphism (Online Resource S2), confirming that this set of markers is suitable for a population genetic study.
Genetic and allelic diversity among populations and years
Genetic and allelic diversity in German populations was similar and relatively high in all years and over all locations. There was no significant difference between years or sample sites by ANOVA after correction according to the Bonferroni method (p > 0.05) (Fig. 2 and Online Resource S4). The mean number of observed alleles (Na) over the loci for each year reached from 6.23 in 2019 to 5.93 in 2017 and was overall similar (ANOVA for years: p = 0.147, for populations: p = 0.414). The mean number of effective alleles (Ne, the number of equally frequent alleles it would take to achieve a given level of gene diversity) was similar in the years 2017, 2018, and 2019, respectively, supporting our findings that there is no significant difference in genetic diversity between years or sample sites (ANOVA for years: p = 0.823, for populations: p = 0.491). Observed heterozygosity (Ho, the observed ratio of heterozygotes) ranged between 0.57 (LB) and 0.71 (KS) in 2019 to values in 2018 between 0.59 in FF to 0.67 in HOH (ANOVA for years: p = 0.313, for populations: p = 0.306). Values for expected heterozygosity (He, the proportion of heterozygous genotypes expected under Hardy–Weinberg equilibrium) were similar in all three years with R showing the overall lowest values in 2017 and 2019 and KS having the highest values during these two years (ANOVA for years: p = 0.566, for populations: p = 0.200). The number of private alleles (an allele that is present in only one population but at any frequency) was low throughout the study period, suggesting that most alleles were shared between populations and years (Fig. 2 and Online Resource S5). The FIS value or inbreeding coefficient of an individual (I) relative to the subpopulation (S) showed a range of -0.07 (R17) to 0.18 (FF18 and LB19) (ANOVA for years: p = 0.203, for populations: p = 0.134). Negative FIS values would reveal a heterozygote excess, while positive values indicate a heterozygote deficit. It was positive for 25 of the German populations, except for R in 2017 ( 0.07) and 2019 ( 0.03) as well as for FF in 2019 ( 0.04), all three showing a heterozygosity excess, which can be a consequence of a genetic bottleneck. In 2018, all populations had a positive FIS value (Fig. 2 and Online Resource S4). Positive values can indicate inbreeding and heterozygote deficiency or to be precise an excess of homozygotes due to the presence of null alleles, inbreeding or population subdivision.

Level of genetic diversity across studied populations over three years. a Na = No. of different alleles; Ne = Number of effective alleles; Ho = observed heterozygosity; He = expected heterozygosity. b AP = No. of private alleles unique to a single population, FIS = mean inbreeding coefficient
Genetic and allelic diversity among laboratory strains
Since the eight laboratory strains were thought to be highly inbred, estimates for genetic and allelic diversity were calculated separately from the German populations to not bias the results. Nevertheless, using laboratory strains as outgroups can provide valuable information, especially for testing the marker system and the effect of artificial breeding in the laboratory. The overall genetic and allelic diversity in the tested laboratory strains was lower than in the field collection from Germany with 42% less different alleles, 38% less effective alleles, 29% less observed heterozygosity and 31% less expected heterozygosity (Fig. 2 and Online Resource S4) making the laboratory strains less diverse than the German field collections. The most diverse laboratory strain was LS_HG18, and the least diverse strain was LS_Canada. These results reflect well the time the strains were kept in the laboratory with LS_HG18 being the newest strain (one year in culture) and LS_Canada being one of the longer established ones (approx. eight years in culture). The FIS value was positive for most strains, which could indicate inbreeding, except for LS_France and LS_Valsugana with negative values, which can be interpreted as an excess of heterozygotes.
Genetic distance and relationship among populations and years
Laboratory strains were excluded from the analysis of molecular variance since they were expected to be highly inbred. AMOVA indicated that 98% of variation occurred within populations, while only 2% appeared among populations in 2017 and 2019. In 2018, 3% of variation originated among populations and 97% from within populations (Table 2). This data suggests that the tested populations were similar to each other and do not show differences between sampling years.
To detect possible genetic relationships between field and laboratory populations, PCoA was performed (Fig. 3). Overall, the separation between German populations was not distinct. The tested laboratory strains were noticeably separated from the German field collections but also from LS_HG18. LS_HG19, on the other hand, those two did show more similarity to the laboratory strains than to the field collections, confirming the impact of laboratory cultivation over time. The laboratory strains showed that the used marker system in our experiment was able to discriminate between populations but that there were no apparent dissimilarities between German sample sites or years. This data suggests that tested German populations were similar over all three years and sample sites, which is in agreement with the results obtained with AMOVA.

PCoA at population level for 2017–2019 generated from 14 microsatellite markers. a Samples of all populations in Germany from 2017 to 2019 (●) and laboratory outgroups (▲ 8- to 10-year-old laboratory strains, ■ laboratory strains that were established between 2014 and 2016, ✖ laboratory strain established from the wild population HG17, ✚ laboratory strains LS_France and LS_Valsugana which were established in the year 2018) b PCoA for German populations sampled in 2017 c The lower left shows the result for the year 2018 d PCoA for German populations sampled in 2019
FST was used as a measure of genetic differentiation between populations. It compares the proportion of the genetic variation contained within a population relative to the total genetic variance and is derived from the variances of allele frequencies. To provide a less biased estimate of gene flow when sample sizes are moderate, FST was estimated instead of RST (Gaggiotti et al. 1999), which is analogous to FST, but is based on the stepwise mutation model (SMM) and can be estimated from the differences of allele sizes. The results revealed a minor differentiation (FST < 0.05) between all populations and years (Fig. 4 and Online Resource S7). In comparison, in experiments including the long-term laboratory strains (LS_USA, LS_Canada, LS_Italy, LS_Frankfurt, LS_France and LS_Valsugana) 54.64% of all FST values showed a moderate (0.1–0.25), 43.17% a minor (FST < 0.1), and 2.19% a strong differentiation (FST > 0.25) (Fig. 4 and Online Resource S7). The pairwise FST for laboratory strain LS_HG revealed that more differentiation was detected in the second year of inbreeding (2019) than in 2018. While the differentiation was moderate in 2018, it was moderate to strong in 2019, which means that the two years of inbreeding resulted in more substantial differentiation compared to its origin (Fig. 4 and Online Resource S7).

Pairwise FST among studied populations over three years. Eight laboratory strains (LS_France, LS_Valsugana, LS_Italy, LS_Frankfurt, LS_USA, LS_Canada, LS_HG18, and LS_HG19) and 28 populations from Germany, collected over three years, are shown in pairwise comparisons. Values are color coded (light gray = FST < 0.1: minor differentiation; gray = 0.1 < FST < 0.25: moderate differentiation; black = FST > 0.25: strong differentiation)
To infer (genetic) population structure, a Bayesian model was used, which detects clusters of genetically similar individuals within subpopulations. First, we excluded all laboratory strains, but STRUCTURE was unable to identify population structure in the German data set. Adding laboratory strains LS_USA, LS_Canada, LS_Italy, LS_France, and LS_Valsugana for comparison revealed two lineages (K = 2) throughout all three years, and populations (Fig. 5). The two most reasonable values for K were K = 2 and K = 3, with the more likely one being K = 2, according to the analysis of ∆K. LS_HG18 and LS_HG19 were excluded from this analysis after we saw that including them changes the STRUCTURE outcome. For the analysis including LS_HG18 and LS_HG19, three lineages (K = 3) were identified, with K = 4 being also a possible K value but not the most likely one, according to the analysis of ∆K. In both cases, the two most likely K are displayed (Fig. 5) to overcome the problem of underestimating the “true” value of K (Janes et al. 2017). These results indicate that German populations are not structured, since only the additional usage of distinct laboratory strains resulted in the detection of population structure. This finding indicates that most likely a single genetic cluster explains the distribution of genetic variation in the sampled German populations.

Structural analysis on D. suzukii populations in Germany. a 34 populations with a total of 670 individuals were analyzed: six laboratory strains (LS_USA, LS_Canada, LS_Italy, LS_Frankfurt, LS_France, and LS_Valsugana), nine populations from 2017, nine populations from 2018 and ten populations from 2019; the possible number of clusters are shown for K = 2 and K = 3. b 36 populations with a total of 710 individuals are shown. In addition to the 34 populations in A), laboratory strains LS_HG18 and LS_HG19 were added to the calculation. Shown are the two clusters K = 3 and K = 4
A neighbor-joining tree based on Nei’s genetic distance corresponds to findings in STRUCTURE and FST (Fig. 6). The German populations were not grouped in any way, and the differences to laboratory strains were visible. While LS_HG18 was already separated from the German field collections after one year in culture, the separation got even more prominent in LS_HG19 after two years in culture.

Unrooted neighbor-joining tree based on Nei‘s genetic distance. Allelic frequencies were obtained with 14 microsatellite markers for the 36 D. suzukii populations. Circles represent population origin with 1 = long-established laboratory strains (2010–2016), 2 = laboratory strains from France (LS_France, 2018) and Italy (LS_Valsugana, 2018), 3 = laboratory strain from Bad Homburg (2017) and 4 = German populations collected over three years (2017–2019) in the field
A bottleneck analysis was used to test the hypothesis of a recent expansion or bottleneck in each of the 28 populations from Germany, not including the laboratory strains since they were irrelevant for the detection of recent reduction or expansion of the population size (Online Resource S9). Under the stepwise mutation model (SMM), no bottleneck event was detected. In contrast, significant heterozygote deficit was identified in FF19 (P = 0.021), PM18 (P = 0.010), R17 (P = 0.002), R18 (P = 0.002) and R19 (P = 0.021) suggesting that a recent expansion took place. However, under the two-phase model (TPM model), none of the abovementioned expansion signals could be confirmed. Instead, a bottleneck event was more likely in HOH18 (P = 0.029), HG18 (P = 0.045) and DO18 (P = 0.045).
Discussion
Invasive species like the SWD constitute a threat to agriculture, economy, and biodiversity (Mooney and Cleland 2001). Thus, understanding population movement, genetic structure, and diversity is one crucial component for the development of pest management strategies. This study addressed the spatial and temporal genetic variation of SWD populations in Germany for three years (2017–2019).
Our analysis of microsatellite markers revealed that levels of genetic diversity in Germany are comparable with other European countries (Tait 2017) and that genetic differentiation of sampled SWD is reflected among individuals within populations but not among populations from different sample sites. None of the populations showed a gain or loss of genetic information over the years. The results suggest a substantial gene flow and a more homogeneous gene pool across different geographical populations. Also, no differences were found between sample sites close to cities like Dortmund (DO) and populations in more rural areas like Derwitz (PM). The mean inbreeding coefficient (FIS) was positive in most populations. This is usually interpreted as a sign of inbreeding, but in this study, precautions were taken during the sampling to avoid this. Another factor that can influence the FIS value is the presence of null alleles. Since null alleles can cause a reduction in heterozygosity, the FIS value increases. However, we did not include microsatellite markers that showed the possibility of null alleles in our final data set.
Furthermore, methodological issues can also be excluded, since the marker system established by Fraimout et al. (2015) and used in this study, was detecting genetic differences between our laboratory strains (LS_USA, LS_Canada, LS_Italy, LS_Frankfurt, LS_Valsugana, LS_France and LS_HG18 or LS_HG19). This study suggests that SWD is a well-established, uniform population in Germany, that might not be altered much by additional invasions. Based on the low differentiation between populations and years, there are either no reinvasions taking place or they do not have an impact on local populations. This is supported by our STRUCTURE, NJ, and PCoA results, which did not group German populations into distinct subpopulations. The lack of admixture suggests that colonization may have involved a single founder population rather than individuals from different origins. This is in contrast to findings from Ukraine, which found evidence for multiple sources of SWD invasions into Europe (Lavrinienko et al. 2017). On the other hand, our results are in accordance with results demonstrating European populations of SWD were genetically more homogenous with lower levels of genetic diversity compared to populations from North America (Adrion et al. 2014; Fraimout et al. 2017).
Based on our presented data, it seems likely that SWD established itself in Germany, which is not a surprise considering its invasion history in other parts of the world (Asplen et al. 2015). SWD has a relatively high reproductive capacity with a single female laying hundreds of eggs during its life with up to 10 generations a year (Walsh et al. 2011), which plays an important role in its invasion success. Additionally, SWD fits the European and German ecosystem, where there is only a constrained number of natural predators and parasitoids (Chabert et al. 2012; Rossi et al. 2015). Geographic barriers like mountains, lakes, and rivers can lead to genetic differences in populations. Germany is only streaked by an orogenic belt of relatively low mountains and hills, the Central German Uplands, but not by high mountain ranges. Based on the low variation among populations, it seems that these geographic properties do not act as barriers or at least they are not isolating populations. Also, SWD can move from high to low elevations, travel long distances by flight and distinct populations from island versus mainland can be found (Tait et al. 2017, 2018). In this respect, it would be interesting to analyze samples from a German island in the North Sea or Baltic Sea in the future. Another factor that can influence populations is the climate. Even though climate, temperature, and humidity differ to a certain extent between sampling locations and years, we were not able to detect these differences in our data. These findings again match the results from Tait et al. (2017), who did not detect differences between the populations from the much colder climate of Trentino compared to the rest of Italy.
All invasive species, independent from their origin, have in common that passive transport enables their successful spread (Banks et al. 2015). Anthropogenic transport of goods is an important aspect for the dissemination of flies since it facilitates the gene flow between locations and international trade via air and sea transport provide new pathways for the spread of insect pest in general (Hulme 2009). Even though it seems to be impossible to reconstruct the exact routes in detail due to the enormous amount of imported fresh produce, it is reasonable to assume that transportation of host fruits and plants lead to an extensive movement of SWD or other pests not only across Germany but all over Europe (Cini et al. 2014). Germany is importing large amounts of host plants and crops from all around the world. A look at the importation routes shows that most fresh fruits are imported from within Europe, namely Spain and Italy but also from the USA, and South America (source: International Trade Center, www.trademap.org). In that respect, distinct populations could be possible, but we did not find proof for this. With the data obtained in this study, it is not possible to reconstruct invasion routes from outside Germany, but would be an interesting aspect for further experiments. However, our collection did not include SWD samples from countries like Spain or the US and, therefore, it is not clear where the German population might have originated from. However, Fraimout et al. (2017) did find evidence that the German sample site in their experiment most likely originated from an admixture of SWD populations from Asia and the eastern US. In general, a better comprehension of genetic structure, population dynamics and the reconstruction of invasion routes could improve the pest control at a regional scale.
Another option for integrated pest management and environmentally friendly alternatives to pesticides could be the Sterile Insect Technique (SIT) that has proven highly effective in agricultural insect species (Krafsur 1998; Benedict and Robinson 2003; Wyss 2006; Augustinos et al. 2017). For SIT programs, sterilized male individuals are released into the environment and lead to infertile matings that could reduce the population size of SWD in the future. Several current SIT programs consider the genetic background refreshing as a vital tool for mating success and efficacy of the release programs (Estes et al. 2012; Zygouridis et al. 2014; Parreño et al. 2014). Due to our findings of genetic uniformity in wild German SWD populations, we hypothesize, that this could be beneficial for the mating success of a single suitable mass-reared SWD strain to different wildtype populations during a SIT program. Therefore, while SWD control is still challenging, biological control methods, including the SIT, remain a beneficial option for sustainable pest control.
Furthermore, our study demonstrates that experiments that are meant to produce data for field applications should be performed with freshly sampled flies because SWD laboratory strains are different from wild populations and can change reasonably within two years. Our newly established laboratory strain LS_HG illustrated the effects of laboratory inbreeding, change of genetic markers, and a decline of allelic diversity. Whether this decline is due to a random selection of individuals during stock keeping or due to laboratory "adaptation" cannot be concluded from our data. However, over two years, the strain got more similar to other laboratory strains. This effect is important to consider during scientific experiments that rely on the use of laboratory strains (Lee et al. 2011; Kinjo et al. 2014; Shearer et al. 2016; Hamby et al. 2016). These strains are often kept over several years or even decades without any changes. This way, transparent and easily reproducible experiments can be performed, and results from those studies are taken as the threshold for similar experiments and as a portrayal of natural processes. However, strains that arise from human cultivation undergo evolutionary genetic changes (Knudsen et al. 2020). Therefore, for evaluations of the efficiency of naturally occurring predators, parasitoids, bacteria or viruses for biological pest control, invasion history, or behavior, fresh field collections or genetically refreshed strains should be the first choice over highly inbred laboratory strains.
Author contributions
SP performed research. SP, SO, and MFS designed experiments, SP, GE, and SO analyzed data and SP, SO, and MFS wrote the paper.
Code availability
Software applications are mentioned in the methods, and programs are publicly available.
References
Adrion JR, Kousathanas A, Pascual M et al (2014) Drosophila suzukii: The genetic footprint of a recent, worldwide invasion. Mol Biol Evol 31:3148–3163. https://doi.org/10.1093/molbev/msu246
Aketarawong N, Chinvinijkul S, Orankanok W et al (2011) The utility of microsatellite DNA markers for the evaluation of area-wide integrated pest management using SIT for the fruit fly, Bactrocera dorsalis (Hendel), control programs in Thailand. Genetica 139:129–140. https://doi.org/10.1007/s10709-010-9510-8
Aketarawong N, Isasawin S, Thanaphum S (2014) Evidence of weak genetic structure and recent gene flow between Bactrocera dorsalis s.s. and B. papaya, across Southern Thailand and West Malaysia, supporting a single target pest for SIT applications. BMC Genet 15:70. https://doi.org/10.1186/1471-2156-15-70
Asplen MK, Anfora G, Biondi A et al (2015) Invasion biology of spotted wing Drosophila (Drosophila suzukii): a global perspective and future priorities. J Pest Sci 88:469–494. https://doi.org/10.1007/s10340-015-0681-z
Augustinos AA, Targovska A, Cancio-Martinez E et al (2017) Ceratitis capitata genetic sexing strains: laboratory evaluation of strains from mass-rearing facilities worldwide. Entomol Exp Appl 164:305–317. https://doi.org/10.1111/eea.12612
Azrag RS, Ibrahim K, Malcolm C et al (2016) Laboratory rearing of Anopheles arabiensis: impact on genetic variability and implications for Sterile Insect Technique (SIT) based mosquito control in northern Sudan. Malar J 15:432. https://doi.org/10.1186/s12936-016-1484-2
Bahder BW, Bahder LD, Hamby KA et al (2015) Microsatellite variation of two Pacific Coast Drosophila suzukii (Diptera: Drosophilidae) Populations. Environ Entomol 44:1449–1453. https://doi.org/10.1093/ee/nvv117
Banks NC, Paini DR, Bayliss KL, Hodda M (2015) The role of global trade and transport network topology in the human-mediated dispersal of alien species. Ecol Lett 18:188–199. https://doi.org/10.1111/ele.12397
Bellamy DE, Sisterson MS, Walse SS (2013) Quantifying host potentials: indexing postharvest fresh fruits for spotted wing Drosophila, Drosophila suzukii. PLoS ONE 8:e61227–e61227. https://doi.org/10.1371/journal.pone.0061227
Benedict MQ, Robinson AS (2003) The first releases of transgenic mosquitoes: an argument for the sterile insect technique. Trends Parasitol 19:349–355. https://doi.org/10.1016/S1471-4922(03)00144-2
Bolda MP, Goodhue RE, Zalom FG (2010) Spotted wing drosophila: potential economic impact of a newly established pest. Agric Resour Econom Update 13:5–8
Boughdad A, Haddi K, El Bouazzati A et al (2021) First record of the invasive spotted wing Drosophila infesting berry crops in Africa. J Pest Sci 94:261–271. https://doi.org/10.1007/s10340-020-01280-0
Briem F, Breuer M, Köppler K, Vogt H (2015) Phenology and occurrence of spotted wing Drosophila in Germany and case studies for its control in berry crops. IOBC-WPRS Bull 109:233–237. http://www.iobcwprs.org/pub/bulletins/bulletin_2015_109_table_of_contents_abstracts.pdf
Briem F, Dominic AR, Golla B et al (2018) Explorative data analysis of Drosophila suzukii trap catches from a seven-year monitoring program in Southwest Germany. Insects 9:125. https://doi.org/10.3390/insects9040125
Calabria G, Máca J, Bächli G et al (2012) First records of the potential pest species Drosophila suzukii (Diptera: Drosophilidae) in Europe. J Appl Entomol 136:139–147. https://doi.org/10.1111/j.1439-0418.2010.01583.x
Chabert S, Allemand R, Poyet M et al (2012) Ability of European parasitoids (Hymenoptera) to control a new invasive Asiatic pest, Drosophila suzukii. Biol Control 63:40–47. https://doi.org/10.1016/j.biocontrol.2012.05.005
Chapman D, Purse BV, Roy HE, Bullock JM (2017) Global trade networks determine the distribution of invasive non-native species. Glob Ecol Biogeogr 26:907–917. https://doi.org/10.1111/geb.12599
Chapman D, Makra L, Albertini R et al (2016) Modelling the introduction and spread of non-native species: international trade and climate change drive ragweed invasion. Glob Chang Biol 22:3067–3079. https://doi.org/10.1111/gcb.13220
Cini A, Anfora G, Escudero-Colomar LA et al (2014) Tracking the invasion of the alien fruit pest Drosophila suzukii in Europe. J Pest Sci 87:559–566. https://doi.org/10.1007/s10340-014-0617-z
Cini A, Ioriatti C, Anfora G (2012) A review of the invasion of Drosophila suzukii in Europe and a draft research agenda for integrated pest management. Bull Insectol 65:149–160
Dalton DT, Walton VM, Shearer PW et al (2011) Laboratory survival of Drosophila suzukii under simulated winter conditions of the Pacific Northwest and seasonal field trapping in five primary regions of small and stone fruit production in the United States. Pest Manag Sci 67:1368–1374. https://doi.org/10.1002/ps.2280
de la Vega GJ, Corley JC, Soliani C (2020) Genetic assessment of the invasion history of Drosophila suzukii in Argentina. J Pest Sci 93:63–75. https://doi.org/10.1007/s10340-019-01149-x
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Estes AM, Nestel D, Belcari A et al (2012) A basis for the renewal of sterile insect technique for the olive fly, Bactrocera oleae (Rossi). J Appl Entomol 136:1–16. https://doi.org/10.1111/j.1439-0418.2011.01620.x
Estoup A, Guillemaud T (2010) Reconstructing routes of invasion using genetic data: Why, how and so what? Mol Ecol 19:4113–4130. https://doi.org/10.1111/j.1365-294X.2010.04773.x
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer H (2010) Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587. https://doi.org/10.1093/genetics/164.4.1567
Food and Agriculture Organization of the United Nations (2021) FAOSTAT. http://www.fao.org/faostat/en/#data/TP. Accessed 15 Feb 2021
Fraimout A, Debat V, Fellous S et al (2017) Deciphering the routes of invasion of Drosophila suzukii by Means of ABC Random Forest. Mol Biol Evol 34:980–996. https://doi.org/10.1093/molbev/msx050
Fraimout A, Loiseau A, Price DK et al (2015) New set of microsatellite markers for the spotted-wing Drosophila suzukii (Diptera: Drosophilidae): a promising molecular tool for inferring the invasion history of this major insect pest. Eur J Entomol 112:855–859. https://doi.org/10.14411/eje.2015.079
Francis RM (2017) pophelper: an R package and web app to analyse and visualize population structure. In: Molecular Ecology Resources. Blackwell Publishing Ltd, pp 27–32. Doi: https://doi.org/10.1111/1755-0998.12509
Gabarra R, Riudavets J, Rodríguez GA et al (2015) Prospects for the biological control of Drosophila suzukii. Biocontrol 60:331–339. https://doi.org/10.1007/s10526-014-9646-z
Gaggiotti OE, Lange O, Rassmann K, Gliddon C (1999) A comparison of two indirect methods for estimating average levels of gene flow using microsatellite data. Mol Ecol 8:1513–1520. https://doi.org/10.1046/j.1365-294x.1999.00730.x
Guo W, Lambertini C, Pyšek P et al (2018) Living in two worlds: evolutionary mechanisms act differently in the native and introduced ranges of an invasive plant. Ecol Evol 8:2440–2452. https://doi.org/10.1002/ece3.3869
Hamby KA, Bolda MP, Sheehan ME, Zalom FG (2014) Seasonal monitoring for Drosophila suzukii (Diptera: Drosophilidae) in California Commercial Raspberries. Environ Entomol 43:1008–1018. https://doi.org/10.1603/EN13245
Hamby KA, Bellamy DE, Chiu JC et al (2016) Biotic and abiotic factors impacting development, behavior, phenology, and reproductive biology of Drosophila suzukii. J Pest Sci 89(3):605–619. https://doi.org/10.1007/s10340-016-0756-5
Hauser M (2011) A historic account of the invasion of Drosophila suzukii (Matsumura) (Diptera: Drosophilidae) in the continental United States, with remarks on their identification. Pest Manag Sci 67:1352–1357. https://doi.org/10.1002/ps.2265
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332. https://doi.org/10.1111/j.1755-0998.2009.02591.x
Hulme PE (2009) Trade, transport and trouble: managing invasive species pathways in an era of globalization. J Appl Ecol 46:10–18. https://doi.org/10.1111/j.1365-2664.2008.01600.x
Ioriatti C, Guzzon R, Anfora G et al (2018) Drosophila suzukii (Diptera: Drosophilidae) contributes to the development of sour rot in grape. J Econ Entomol 111:283–292. https://doi.org/10.1093/jee/tox292
Jakobs R, Gariepy TD, Sinclair BJ (2015) Adult plasticity of cold tolerance in a continental-temperate population of Drosophila suzukii. J Insect Physiol 79:1–9. https://doi.org/10.1016/j.jinsphys.2015.05.003
Janes JK, Miller JM, Dupuis JR et al (2017) The K = 2 conundrum. Mol Ecol. https://doi.org/10.1111/mec.14187
Jarne P, Lagoda PJL (1996) Microsatellites, from molecules to populations and back. Trends Ecol Evol 11:424–429. https://doi.org/10.1016/0169-5347(96)10049-5
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106. https://doi.org/10.1111/j.1365-294X.2007.03089.x
Kinjo H, Kunimi Y, Nakai M (2014) Effects of temperature on the reproduction and development of Drosophila suzukii (Diptera: Drosophilidae). Appl Entomol Zool 49:297–304. https://doi.org/10.1007/s13355-014-0249-z
Knudsen KE, Reid WR, Barbour TM et al (2020) Genetic variation and potential for resistance development to the TTA overexpression lethal system in insects. G3 Genes Genomes Genetics 10:1271–1281. https://doi.org/10.1534/g3.120.400990
Krafsur ES (1998) Sterile insect technique for suppressing and eradicating insect population: 55 years and counting. Entomology Publications, p 417. https://lib.dr.iastate.edu/ent_pubs/417
Lanzavecchia SB, Juri M, Bonomi A et al (2014) Microsatellite markers from the “South American fruit fly” Anastrepha fraterculus: a valuable tool for population genetic analysis and SIT applications. BMC Genet 15:S13. https://doi.org/10.1186/1471-2156-15-S2-S13
Lavrinienko A, Kesäniemi J, Watts PC et al (2017) First record of the invasive pest Drosophila suzukii in Ukraine indicates multiple sources of invasion. J Pest Sci 90(2):421–429. https://doi.org/10.1007/s10340-016-0810-3
Lee JC, Bruck DJ, Curry H et al (2011) The susceptibility of small fruits and cherries to the spotted-wing drosophila, Drosophila suzukii. Pest Manag Sci 67:1358–1367. https://doi.org/10.1002/ps.2225
Lee KZ, Vilcinskas A (2017) Analysis of virus susceptibility in the invasive insect pest Drosophila suzukii. J Invertebr Pathol 148:138–141. https://doi.org/10.1016/j.jip.2017.06.010
Matsumura S (1931) 6000 illustrated insects of Japan-Empire (in Japanese). Tokohshoin, Tokyo, p 1497
Mazzi D, Bravin E, Meraner M et al (2017) Economic impact of the introduction and establishment of Drosophila suzukii on sweet cherry production in Switzerland. Insects 8:18. https://doi.org/10.3390/insects8010018
Mooney HA, Cleland EE (2001) The evolutionary impact of invasive species. Proc Natl Acad Sci U S A 98:5446–5451. https://doi.org/10.1073/pnas.091093398
Nei M, Tajima F, Tateno Y (1983) Accuracy of estimated phylogenetic trees from molecular data—II. Gene frequency data. J Mol Evol 19:153–170. https://doi.org/10.1007/BF02300753
Nei M, Takezaki N (1983) Estimation of genetic distances and phylogenetic trees from DNA analysis. Proc 5th World Cong Genet Appl Livstock Prod 21:405–412
Paetkau D, Slade R, Burden M, Estoup A (2004) Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65. https://doi.org/10.1046/j.1365-294X.2004.02008.x
Parreño MA, Scannapieco AC, Remis MI et al (2014) Dynamics of genetic variability in Anastrepha fraterculus (Diptera: Tephritidae) during adaptation to laboratory rearing conditions. BMC Genet 15:S14. https://doi.org/10.1186/1471-2156-15-S2-S14
Peakall R, Smouse PE (2012) GenALEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Piry S, Luikart GCJ (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503. https://doi.org/10.1093/jhered/90.4.502.
Piry S, Alapetite A, Cornuet JM et al (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539. https://doi.org/10.1093/jhered/esh074
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1534/genetics.116.195164
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci U S A 94:9197–9201. https://doi.org/10.1073/pnas.94.17.9197
Renkema JM, Telfer Z, Gariepy T, Hallett RH (2015) Dalotia coriaria as a predator of Drosophila suzukii: Functional responses, reduced fruit infestation and molecular diagnostics. Biol Control 89:1–10. https://doi.org/10.1016/j.biocontrol.2015.04.024
Rombaut A, Guilhot R, Xuéreb A et al (2017) Invasive Drosophila suzukii facilitates Drosophila melanogaster infestation and sour rot outbreaks in the vineyards. R Soc Open Sci 4:170117. https://doi.org/10.1098/rsos.170117
Rossi Stacconi MV, Buffington M, Daane KM et al (2015) Host stage preference, efficacy and fecundity of parasitoids attacking Drosophila suzukii in newly invaded areas. Biol Control 84:28–35. https://doi.org/10.1016/j.biocontrol.2015.02.003
Rota-Stabelli O, Ometto L, Tait G et al (2020) Distinct genotypes and phenotypes in European and American strains of Drosophila suzukii: implications for biology and management of an invasive organism. J Pest Sci 93(1):77–89. https://doi.org/10.1007/s10340-019-01172-y
Rout TM, Moore JL, Possingham HP, McCarthy MA (2011) Allocating biosecurity resources between preventing, detecting, and eradicating island invasions. Ecol Econ 71:54–62. https://doi.org/10.1016/j.ecolecon.2011.09.009
Schlötterer C (2000) Evolutionary dynamics of microsatellite DNA. Chromosoma 109:365–371. https://doi.org/10.1007/s004120000089
Schlötterer C (2004) The evolution of molecular markers—Just a matter of fashion? Nat Rev Genet 5:63–69. https://doi.org/10.1038/nrg1249
Schrader L, Kim JW, Ence D et al (2014) Transposable element islands facilitate adaptation to novel environments in an invasive species. Nat Commun 5:1–10. https://doi.org/10.1038/ncomms6495
Selkoe KA, Toonen RJ (2006) Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett 9:615–629. https://doi.org/10.1111/j.1461-0248.2006.00889.x
Shearer PW, West JD, Walton VM et al (2016) Seasonal cues induce phenotypic plasticity of Drosophila suzukii to enhance winter survival. BMC Ecol 16:1–18. https://doi.org/10.1186/s12898-016-0070-3
Shorthouse DP (2010) SimpleMappr, an online tool to produce publication-quality point maps. http://www.simplemappr.net. Accessed 18 July 2020.
Stephens AR, Asplen MK, Hutchison WD, Venette RC (2015) Cold Hardiness of Winter-Acclimated Drosophila suzukii (Diptera: Drosophilidae) Adults. Environ Entomol 44:1619–1626. https://doi.org/10.1093/ee/nvv134
Stockton DG, Wallingford AK, Brind’amore G et al (2020) Seasonal polyphenism of spotted-wing Drosophila is affected by variation in local abiotic conditions within its invaded range, likely influencing survival and regional population dynamics. Ecol Evol 10:7669–7685. https://doi.org/10.1002/ece3.6491
Tait G, Grassi A, Pfab F et al (2018) Large-scale spatial dynamics of Drosophila suzukii in Trentino, Italy. J Pest Sci 91:1213–1224. https://doi.org/10.1007/s10340-018-0985-x
Tait G, Vezzulli S, Sassù F et al (2017) Genetic variability in Italian populations of Drosophila suzukii. BMC Genet 18:87. https://doi.org/10.1186/s12863-017-0558-7
Takezaki N, Nei M, Tamura K (2014) POPTREEW: web version of POPTREE for constructing population trees from Allele frequency data and computing some other quantities. Mol Biol Evol 31:1622–1624. https://doi.org/10.1093/molbev/msu093
Vogt H, Baufeld P, Gross J et al (2012) Drosophila suzukii: a new threat feature for the European fruit and viticulture-report for the international conference in Trient, 2, December 2011. J für Kulturpflanzen 64:68–72
Walsh DB, Bolda MP, Goodhue RE et al (2011) Drosophila suzukii (Diptera: Drosophilidae): invasive pest of ripening soft fruit expanding its geographic range and damage potential. J Integr Pest Manag 2:G1–G7. https://doi.org/10.1603/ipm10010
Wyss JH (2006) Screwworm eradication in the Americas. Ann N Y Acad Sci 916:186–193. https://doi.org/10.1111/j.1749-6632.2000.tb05289.x
Xia L, Geng Q, An S (2020) Rapid genetic divergence of an invasive species, Spartina alterniflora, in China. Front Genet 11:284. https://doi.org/10.3389/fgene.2020.00284
Zygouridis NE, Argov Y, Nemny-Lavy E et al (2014) Genetic changes during laboratory domestication of an olive fly SIT strain. J Appl Entomol 138:423–432. https://doi.org/10.1111/jen.12042
Acknowledgements
We thank Tanja Rehling for technical assistance on molecular assays. Special thanks goes to Dr. Eric Marois, Dr. Alberto Grassi, Dr. Ruth Jakobs and Dr. Kwang-Zin Lee for providing us with samples from Strasbourg, Trentino, Ontario and Frankfurt, respectively. We also want to thank Dr. Felix Briem and Dr. Heidrun Vogt from the JKI Dossenheim, Dr. Michael Breuer from the WBI Freiburg, Mrs. Ulrike Holz from the LELF Brandenburg and Mr. Karlheinz Geipel from the LFL Bayern for sending us D. suzukii samples. Further, we want to thank Irina Häcker, Roswitha Aumann, Moni Gerl, Angela and Erwin Hobein, Franziska Scholpp, Laura Thies, Ute Lugert, Christine Best, Thomas Steltner and Tobias Lautwein for sending us samples as well.
Funding
Open Access funding enabled and organized by Projekt DEAL.. The research was funded by the Faculty of Agricultural Sciences, Nutritional Sciences, and Environmental Management, FB09, through the professorship of Insect Biotechnology and Plant Protection of the JLU Gießen.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or vertebrates performed by any of the authors.
Consent for publication
We consent with the publication of all submitted data.
Availability of data and material
All data discussed in the paper will be made available to readers.
Additional information
Communicated by Antonio Biondi.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Petermann, S., Otto, S., Eichner, G. et al. Spatial and temporal genetic variation of Drosophila suzukii in Germany. J Pest Sci 94, 1291–1305 (2021). https://doi.org/10.1007/s10340-021-01356-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10340-021-01356-5