
Abstract
We find polymers everywhere in our daily activities, for example, as a part of consumer electronics products, healthcare devices, vehicles, etc. Analytical characterization of such materials is an important step towards understanding their properties and behavior in various applications. The increase of material complexity driven by highly demanding requirements for many applications necessitates the use of sophisticated analytical techniques to obtain sufficient insight into the structure of these materials. Coupling of liquid chromatography with other information-rich instrumental techniques becomes more and more important in the field of polymer characterization. Such combination can enable simultaneous separation, identification, and quantification of polymer sample components. In addition, it can provide information on interdependence of two polymer properties, e.g., molecular weight and chemical composition. Different hyphenated systems may be applied to address different problems in polymer research and development and a selection of the right technique may not be an easy and straightforward task. In this paper, the applications of LC-NMR, LC-IR, LC-Raman, LC-MS, LC-MALDI, LC × LC, and LC × Py-GC for polymer analysis are reviewed, their advantages and limitations are discussed, and practical challenges for the implementation of these techniques in a lab are addressed. Different hyphenated options are compared to facilitate selection of a suitable instrument for the particular problem at hand.

Reproduced by permission of the Royal Society of Chemistry


Reprinted with permission from [49]. Copyright 2010 American Chemical Society

Reprinted with permission from [51]. Copyright 2010 American Chemical Society

Reprinted with permission from [88]. Copyright 2011 American Chemical Society


Reprinted from [92], with permission from Elsevier

Reprinted with permission from [94]. Copyright 2013 American Chemical Society

Reprinted with permission from [97]. Copyright 2012 American Chemical Society
Similar content being viewed by others
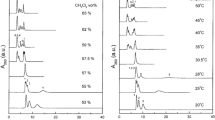

References
Pasch H, Trathnigg B (2013) Multidimensional HPLC of polymers. Springer, Berlin, Heidelberg
Pasch H (2013) Hyphenated separation techniques for complex polymers. Polym Chem 4:2628–2650
Malik MI, Pasch H (2014) Novel developments in the multidimensional characterization of segmented copolymers. Prog Polym Sci 39:87–123
Stuart B (2004) Infrared spectroscopy: fundamentals and applications. Wiley, Chichester
Fuller MP, Griffiths PR (1978) Diffuse reflectance measurements by infrared Fourier transform spectrometry. Anal Chem 50:1906–1910
Vidrine DW, Mattson DR (1978) A practical real-time Fourier transform infrared detector for liquid chromatography. Appl Spectrosc 32:502–506
Plass M, Albrecht A, Bruell R (2010) Liquid Chromatography infrared and Size Exclusion Chromatography-infrared Analysis for Polymer Characterization. In: Encyclopedia of analytical chemistry. Wiley, Weinheim
Kok SJ, Wold CA, Hankemeier Th, Schoenmakers PJ (2003) Comparison of on-line flow-cell and off-line solvent-elimination interfaces for size-exclusion chromatography and Fourier-transform infrared spectroscopy in polymer analysis. J Chromatogr A 1017:83–96
Pasch H, Malik MI, Macko T (2013) Recent advances in high-temperature fractionation of polyolefins. Adv Polym Sci 251:77–140
Kuligowski J (2011) New instrumental and chemometric developments for the on-line hyphenation of liquid chromatography and infrared spectroscopy. Univ. Valencia, Valencia
Kuligowski J (2012) On-line coupling of liquid chromatography and infrared spectroscopy: new instrumental and chemometric tools LAP LAMBERT. Academic Publishing, Saarbrücken
Istvan K, Rajko R, Keresztury G (2006) Towards the solution of the eluent elimination problem in high-performance liquid chromatography–infrared spectroscopy measurements by chemometric methods. J Chromatogr A 1104:154–163
Kuligowski J, Quintás G, Garrigues S, Lendl B, de la Guardia M (2010) Recent advances in on-line liquid chromatography-infrared spectrometry (LC-IR). TrAC Trends Anal Chem 29:544–552
Kuligowski J, Quintás G, Garrigues S, de la Guardia M (2010) Application of point-to-point matching algorithms for background correction in on-line liquid chromatography–Fourier transform infrared spectrometry (LC–FTIR). Talanta 80:1771–1776
Kuligowski J, Quintás G, Garrigues S, de la Guardia M (2009) New background correction approach based on polynomial regressions for on-line liquid chromatography–Fourier transform infrared spectrometry. J Chromatogr A 1216:3122–3130
Kuligowski J, Quintás G, Tauler R, Lendl B, De la Guardia M (2011) Background correction and multivariate curve resolution of online liquid chromatography with infrared spectrometric detection. Anal Chem 83:4855–4862
Johnson CC, Hellgeth JW, Taylor LT (1985) Reversed-phase liquid chromatography with Fourier infrared spectrometric detection using a flow cell interface. Anal Chem 57:610–615
Somsen GW, Gooijer C, Brinkman UATh (1999) Liquid chromatography—Fourier-transform infrared spectrometry. J Chromatogr A 856:213–242
Willis JN, Dwyer JL, Liu MX (1997) Polymer characterization using SEC-FTIR. Int J Polym Anal Charact 4:21–29
Kuligowski J, Quintás G, De la Guardia M, Lendl B (2010) Analytical potential of mid-infrared detection in capillary electrophoresis and liquid chromatography: a review. Anal Chim Acta 679:31–42
Dwyer JL, Zhou M (2011) Polymer characterization by combined chromatography-infrared spectroscopy. Int J Spectrosc 2011:1–12
Jansen JAJ (1990) On-line liquid chromatography—Fourier transform infrared spectroscopy for the analysis of polymers and additives. Fresenius J Anal Chem 337:398–402
Zhou M (2011) LC-IR applications in polymer industries. http://www.slideshare.net/mzhou45/lcir-applications-in-polymer-related-industries. Accessed Jan 2017
Prabhu KN, Macko T, Bruell R, Remerie K, Tacx J, Garg P, Ginzburg A (2016) Separation of maleic anhydride grafted polypropylene using multidimensional high-temperature liquid chromatography. J Chromatogr A 1441:96–105
Malanin M, Eichhorn KJ, Lederer A, Treppe P, Adam G, Fischer D, Voigt D (2009) On-line preferential solvation studies of polymers by coupled chromatographic-Fourier transform infrared spectroscopic flow-cell technique. J Chromatogr A 1216:8939–8946
Piel C, Albrecht A, Neubauer C, Klampfl CW, Reussner J (2011) Improved SEC-FTIR method for the characterization of multimodal high-density polyethylenes. Anal Bioanal Chem 400:2607–2613
Beskers TF, Hofe T, Wilhelm M (2015) Development of a chemically sensitive online SEC detector based on FTIR spectroscopy. Polym Chem 6:128–142
Beskers TF, Brandstetter M, Kuligowski J, Quintas G, Wilhelm M, Lendl B (2014) High performance liquid chromatography with mid-infrared detection based on a broadly tunable quantum cascade laser. Analyst 139:2057–2064
Schluecker S (2010) Surface enhanced Raman spectroscopy analytical, biophysical and life science applications. Wiley-VCH, Weinheim
Hyphenated and alternative methods of detection in chromatography. CRC Press, Taylor & Francis group, Boca Raton. 2012
Dijkstra RJ, Ariese F, Gooijer C, Brinkman UATh (2005) Raman spectroscopy as a detection method for liquid-separation techniques. TrAC Trends Anal Chem 24:304–323
Pothier NJ, Force RK (1990) Surface-enhanced Raman spectroscopy at a silver electrode as a detection system in flowing streams. Anal Chem 62:678–680
Pothier NJ, Force RK (1994) Detection of biologically important compounds in flowing aqueous streams by surface-enhanced Raman spectroscopy at a silver electrode. Appl Spectrosc 48:421–425
Sheng R, Ni F, Cotton TM (1991) Determination of purine bases by reversed-phase high-performance liquid chromatography using real-time surface-enhanced Raman spectroscopy. Anal Chem 63:437
Somsen GW, Coulter SK, Gooijer C, Velthorst NH, Brinkman UATh (1997) Coupling of column liquid chromatography and surface-enhanced resonance Raman spectroscopy via a thin-layer chromatographic plate. Anal Chim Acta 349:189–197
Seifar RM, Altelaar MAF, Dijkstra RJ, Ariese F, Brinkman UATh, Gooijer C (2000) Surface-enhanced resonance Raman spectroscopy as an identification tool in column liquid chromatography. Anal Chem. 72:5718–5724
Farquharson S, Maksymiuk P. Simultaneous chemical separation and surface-enhanced raman spectral detection using metal-doped sol-gels. US Patent US 6943031 B22005
Dijkstra RJ, Bader AN, Hoornweg G, Hoornweg GPh, Brinkman UATh, Gooijer C (1999) On-line coupling of column liquid chromatography and Raman spectroscopy using a liquid core waveguide. Anal Chem 71:4575
Surowiec I, Baena JR, Frank J, Laurell Th, Nilsson J, Trojanowicz M, Lendl B (2005) Flow-through microdispenser for interfacing μ-HPLC to Raman and mid-IR spectroscopic detection. J Chromatogr A 1080:132–139
Pitkanen L, Urbas AA, Striegel AM (2015) On the feasibility of determining polymer chemical heterogeneity by SEC with continuous off-line Raman detection. Polym Chem 6:4864–4874
Hatada K, Kitayama T (2004) NMR spectroscopy of polymers. Springer, Berlin
Albert K (2002) On-line LC-NMR and related techniques, 1st edn. Wiley, Chichester
Elipe MVS (2011) LC-NMR and other hyphenated NMR techniques, 1st edn. Wiley, Hoboken
Gonnella NC (2013) LC-NMR. Expanding the limits of structure elucidation. CRC Press, Boca Raton
Cladridge TWD (1999) High-resolution NMR techniques in organic chemistry. Elsevier, Amsterdam
Hiller W, Sinha P, Hehn M, Pasch H (2014) On-line LC-NMR—from an expensive toy to a powerful tool in polymer analysis. Prog Polym Sci 29:979–1016
Hiller W, Hehn M, Hofe T, Oleschko K, Montag P (2012) On-line fractionated size exclusion chromatography-nuclear magnetic resonance of polymers with 1H and 2H nuclear magnetic resonance detection. J Chromatogr A 1240:77–82
Sturm S, Seger C (2012) Liquid chromatography–nuclear magnetic resonance coupling as alternative to liquid chromatography–mass spectrometry hyphenations: curious option or powerful and complementary routine tool? J Chromatogr A 1259:50–61
Hiller W, Hehn M, Hofe T, Oleschko K (2010) Online size exclusion chromatography–NMR for the determination of molar mass distributions of copolymers. Anal Chem 82:8244–8250
Hehn M, Wagner T, Hiller W (2014) Direct quantification of molar masses of copolymers by online liquid chromatography under critical conditions-nuclear magnetic resonance and size exclusion chromatography–nuclear magnetic resonance. Anal Chem 86:490–497
Hiller W, Pasch H, Sinha P, Wagner T, Thiel J, Wagner M et al (2010) Coupling of NMR and liquid chromatography at critical conditions: a new tool for the block length and microstructure analysis of block copolymers. Macromolecules 43:4853–4863
Hehn M, Sinha P, Pasch H, Hiller W (2015) Onflow liquid chromatography at critical conditions coupled to 1H and 2H nuclear magnetic resonance as powerful tools for the separation of poly(methylmethacrylate) according to isotopic composition. J Chromatogr A 1387:69–74
Hehn M, Maiko K, Pasch H, Hiller W (2013) An efficient method for the analysis of PMMA with respect to tacticity. Macromolecules 46:7678–7686
Cudaj M, Guthausen G, Hofe T, Wilhelm M (2011) SEC-MR-NMR: online coupling of size exclusion chromatography and medium resolution NMR spectroscopy. Macromol Rapid Commun 32:665–670
Holcapek M, Jirasko R, Lisa M (2012) Recent developments in liquid chromatography–mass spectrometry and related techniques. J Chromatogr A 1259:3–15
Hoteling AJ, Papagelis PT (2014) Structural characterization of silicon polymers using compositional ultra-high performance liquid chromatography separation, electrospray ionization, and high resolution/accurate mass. Anal Chim Acta 808:231–239
Gruendling T, Guilhaus M, Barner-Kowollik C (2009) Fast and accurate determination of absolute individual molecular weight distributions from mixtures of polymers via size exclusion chromatography–electrospray ionization mass spectrometry. Macromolecules 42:6366–6374
Gruendling T, Junkers T, Guilhaus M, Barner-Kowollik C (2010) Mark-Houwink parameters for the universal calibration of acrylate, methacrylate and vinyl acetate polymers determined by online size-exclusion chromatography–mass spectrometry. Macromol Chem Phys 211:520–528
Falkenhagen J, Weidner S (2009) Determination of critical conditions of adsorption for chromatography of polymers. Anal Chem 81:282–287
Schneider C, Sablier M, Desmazieres B (2008) Characterization by mass spectrometry of an unknown polysiloxane sample used under uncontrolled medical conditions for cosmetic surgery. Rapid Commun Mass Spec. 22:3353–3361
van Leeuwen SM, Tan B, Grijpma DW, Feijen J, Karst U (2007) Characterization of the chemical composition of a block copolymer by liquid chromatography/mass spectrometry using atmospheric pressure chemical ionization and electrospray ionization. Rapid Commun Mass Spec 21:2629–2637
Crotty S, Gerislioglu S, Endres KJ, Wesdemiotis C, Schubert US (2016) Polymer architecture via mass spectrometry and hyphenated techniques: a review. Anal Chim Acta 932:1–21
Barner-Kowollik C, Gruendling T, Falkenhagen J, Weidner S (2012) Mass spectrometry in polymer chemistry. Wiley VCH, Weinheim
Montaudo G, Samperi F, Montaudo M (2006) Characterization of synthetic polymers by MALDI–MS. Prog Polym Sci 31:277–357
Murgasova R, Hercules DM (2003) MALDI of synthetic polymers—an update. Int J Mass Spectrom 226:151–162
Musyimi HK, Narcisse DA, Zhang X, Stryjewski W, Soper SA, Murray KK (2004) On-line CE-MALDI TOF MS using a rotating ball interface. Anal Chem 76:5968–5973
Preisler J, Hu P, Rejtar T, Karger BL (2000) Capillary electrophoresis-matrix assisted laser desorption/ionization time-of-flight mass spectrometry using a vacuum deposition interface. Anal Chem 72:4785–4795
Orsnes H, Graf T, Degn H, Murray KK (2000) A rotating ball inlet for on-line MALDI mass spectrometry. Anal Chem 72:251–254
Fei X, Wei G, Murray KK (1996) On-line coupling of gel permeation chromatography with aerosol MALDI mass spectrometry. Anal Chem 68:3555–3560
Zhan Q, Gusev A, Hercules DM (1999) A novel interface for on-line coupling of liquid capillary chromatography with matrix-assisted laser desorption/ionization detection. Rapid Commun Mass Spec 13:2278–2283
Laiko VV, Baldwin MA, Burlingame AL (2000) Atmospheric pressure matrix-assisted laser desorption/ionization mass spectrometry. Anal Chem 72:652–657
Daniel JM, Laiko VV, Doroshenko VM, Zenobi R (2005) Interfacing liquid chromatography with atmospheric pressure MALDI-MS. Anal Bioanal Chem 383:895–902
Daniel JM, Ehala S, Friess SD, Zenobi R (2004) On-line atmospheric pressure matrix-assisted laser desorption/ionization mass spectrometry. Analyst 129:574–578
Li L (2010) MALDI mass spectrometry for synthetic polymer analysis. Wiley, Hoboken
Zhang BY, McDonald C, Li L (2004) Combining liquid chromatography with MALDI mass spectrometry using a heated droplet interface. Anal Chem 76:992–1001
Young JB, Li L (2007) Impulse-driven heated-droplet deposition interface for capillary and microbore LC-MALDI MS and MS/MS. Anal Chem 79:5927–5934
Weidner S, Falkenhagen J (2009) Imaging mass spectrometry for examining localization of polymeric composition in matrix-assisted laser desorption/ionization samples. Rapid Commun Mass Spec 23:653–660
Weidner S, Knappe P, Panne U (2011) MALDI-TOF imaging mass spectrometry of artifacts in “dried droplet” polymer samples. Anal Bioanal Chem 401:127–134
Gabriel SJ, Schwarzinger C, Schwarzinger B, Panne U, Weidner S (2014) Matrix segregation as the major cause for sample inhomogeneity in MALDI dried droplet spots. J Am Soc Mass Spectrom 25:1356–1363
Coulier L, Kaal ER, Hankemeier T (2005) Comprehensive two-dimensional liquid chromatography and hyphenated liquid chromatography to study the degradation of poly(bisphenol A)carbonate. J Chromatogr A 1070:79–87
Esser E, Keil C, Braun D, Montag P, Pasch H (2000) Matrix-assisted laser desorption/ionization mass spectrometry of synthetic polymers. 4. Coupling of size exclusion chromatography and MALDI-TOF using a spray-deposition interface. Polymer 41:4039–4046
Falkenhagen J, Friedrich JF, Schulz G, Kruger RP, Much H, Weidner S (2000) Liquid adsorption chromatography near critical conditions of adsorption coupled with matrix-assisted laser desorption/ionization mass spectrometry. Int J Polym Anal Charact 5:549–562
Lou XW, van Dongen JLJ (2000) Direct sample fraction deposition using electrospray in narrow-bore size-exclusion chromatography/matrix-assisted laser desorption/ionization time-of-flight mass spectrometry for polymer characterization. J Mass Spectrom 35:1308–1312
Axelsson J, Hoberg AM, Waterson C, Myatt P, Shield GL, Varney J et al (1997) Improved reproducibility and increased signal intensity in matrix-assisted laser desorption/ionization as a result of electrospray sample preparation. Rapid Commun Mass Spec 11:209–213
Hanton SD, Hyder IZ, Stets JR, Owens KG, Blair WR, Guttman CM et al (2004) Investigations of electrospray sample deposition for polymer MALDI mass spectrometry. J Am Soc Mass Spectrom 15:168–179
Nielen MWF (1998) Polymer analysis by micro-scale size-exclusion chromatography/MALDI time-of-flight mass spectrometry with a robotic interface. Anal Chem 70:1563–1568
Weidner S, Falkenhagen J, Krueger R-P, Just U (2007) Principle of two-dimensional characterization of copolymer. Anal Chem 79:4814–4819
Weidner S, Falkenhagen J (2011) LC-MALDI-TOF imaging MS: a new approach in combining chromatography and mass spectrometry of copolymers. Anal Chem 83:9153–9158
Pasch H, Schrepp W (2003) MALDI-TOF mass spectrometry of synthetic polymers. Springer, Berlin
Baumgaertel A, Altuntas E, Schubert US (2012) Recent developments in the detailed characterization of polymers by multidimensional chromatography. J Chromatogr A 1240:1–20
Uliyanchenko E, van der Wal S, Schoenmakers PJ (2012) Challenges in polymer analysis by liquid chromatography. Polym Chem 3:2313–2335
Malik MI, Lee S, Chang T (2016) Comprehensive two-dimensional liquid chromatographic analysis of poloxamers. J Chromatogr A 1442:33–41
Jeong J, Kim K, Lee R, Lee S, Kim H, Jung H et al (2014) Preparation and analysis of bicyclic polystyrene. Macromolecules 47:3791–3796
Maiko K, Hehn M, Hiller W, Pasch H (2013) Comprehensive two-dimensional liquid chromatography of stereoregular poly(methyl methacrylates) for tacticity and molar mass analysis. Anal Chem 85:9793–9798
Prabhu KN, Bruell R, Macko T, Remerie K, Tacx J, Garg P, Ginzburg A (2015) Separation of bimodal high density polyethylene using multidimensional high temperature liquid chromatography. J Chromatogr A 1419:67–80
MacNair JE, Lewis KC, Jorgenson JW (1997) Ultrahigh-pressure reversed-phase liquid chromatography in packed capillary columns. Anal Chem 69:983–989
Uliyanchenko E, Cools PJCH, van der Wal S, Schoenmakers PJ (2012) Comprehensive two-dimensional ultrahigh-pressure liquid chromatography for separations of polymers. Anal Chem 84:7802–7809
Janco M, Alexander JN, Bouvier ESP, Morrison D (2013) Ultra-high performance size-exclusion chromatography of synthetic polymers. J Sep Sci 36:2718–2727
Uliyanchenko E, Wold C (2016) Ultrahigh-pressure size-exclusion separations of engineering plastics: challenges and opportunities. LCGC Europe 29:22–27
Li JW, Carr PW (1997) Accuracy of empirical correlations for estimating diffusion coefficients in aqueous organic mixtures. Anal Chem 69:2530–2536
Anita FD, Horvath C (1988) High-performance liquid chromatography at elevated temperatures: examination of conditions for the rapid separation of large molecules. J Chromatogr A 435:1–15
Stoll DR, Li X, Wang X, Carr PW, Porter SEG, Rutan SC (2007) Fast, comprehensive two-dimensional liquid chromatography. J Chromatogr A 1168:3–43
Im K, Park HW, Lee S, Chang T (2009) Two-dimensional liquid chromatography analysis of synthetic polymers using fast size exclusion chromatography at high column temperature. J Chromatogr A 1216:4606–4610
Hiller W, Hehn M, Sinha P, Raust J-A, Pasch H (2012) Online coupling of two-dimensional liquid chromatography and NMR for the analysis of complex polymers. Macromolecules 45:7740–7748
Kok SJ, Hankemeier Th, Schoenmakers PJ (2005) Comprehensive two-dimensional liquid chromatography with on-line Fourier-transform-infrared-spectroscopy detection for the characterization of copolymers. J Chromatogr A 1098:104–110
Barqawi H, Ostas E, Liu B, Carpentier J-F, Binder W (2012) Multidimensional charaterization of alpha, omega-telechelic poly(epsilon-caprolactone)s via online coupling of 2D chromatographic methods (LC/SEC) and ESI-TOF/MALDI-TOF-MS. Macromolecules 45:9779–9790
Kaal ER, Alkema G, Kurano M, Geissler M, Janssen H-G (2007) On-line size exclusion chromatography–pyrolysis-gas chromatography–mass spectrometry for copolymer characterization and additive analysis. J Chromatogr A 1143:182–189
Kaal ER, Kurano M, Geissler M, Janssen H-G (2008) Hyphenation of aqueous liquid chromatography to pyrolysis-gas chromatography and mass spectrometry for the comprehensive characterization of water-soluble polymers. J Chromatogr A 1186:222–227
Brander E, Wold C (2014) The identification and quantification of a high molecular-weight light stabilizer in polycarbonate by application of an online coupling of size exclusion chromatography in stopped flow mode with pyrolysis gas chromatography time of flight mass spectroscopy. J Chromatogr A 1362:309–312
Kaal ER (2010) Extending the applicability of gas chromatography. University of Amsterdam, Amsterdam
Acknowledgements
The author would like to acknowledge Christian Wold and Olivier Guise for their valuable suggestions on the content of this manuscript. The author also wants to thank Johannes Guenther for the useful discussions on the LC-NMR hyphenation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that she has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by the author.
Additional information
Published in the topical collection Young Investigators in Separation Science with editors D. Mangelings, G. Massolini, G. K. E. Scriba, R. M. Smith, and A. M. Striegel.
Rights and permissions
About this article

Cite this article
Uliyanchenko, E. Applications of Hyphenated Liquid Chromatography Techniques for Polymer Analysis. Chromatographia 80, 731–750 (2017). https://doi.org/10.1007/s10337-016-3193-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-016-3193-y