
Abstract
Respiratory viruses are opportunistic pathogens that infect the upper respiratory tract in humans and cause severe illnesses, especially in vulnerable populations. Some viruses have neuroinvasive properties and activate the immune response in the brain. These immune events may be neuroprotective or they may cause long-term damage similar to what is seen in some neurodegenerative diseases. The new “Severe Acute Respiratory Syndrome Coronavirus 2” (SARS-CoV-2) is one of the Respiratory viruses causing highly acute lethal pneumonia coronavirus disease 2019 (COVID-19) with clinical similarities to those reported in “Severe Acute Respiratory Syndrome Coronavirus”(SARS-CoV) and the “Middle East Respiratory Syndrome Coronavirus”(MERS-CoV) including neurological manifestation. To examine the possible neurological damage induced by SARS-CoV-2, it is necessary to understand the immune reactions to viral infection in the brain, and their short- and long-term consequences. Considering the similarities between SARS-CoV and SARS-CoV-2, which will be discussed, cooperative homological and phylogenetical studies lead us to question if SARS-CoV-2 can have similar neuroinvasive capacities and neuroinflammatiory events that may lead to the same short- and long-term neuropathologies that SARS-CoV had shown in human and animal models. To explain the neurological manifestation caused by SARS-CoV-2, we will present a literature review of 765 COVID-19 patients, in which 18% had neurological symptoms and complications, including encephalopathy, encephalitis and cerebrovascular pathologies, acute myelitis, and Guillain-Barré syndrome. Clinical studies describe anosmia or partial loss of the sense of smell as the most frequent symptom in COVID19 patients, suggesting that olfactory dysfunction and the initial ultrarapid immune responses could be a prognostic factor.
Similar content being viewed by others


Introduction
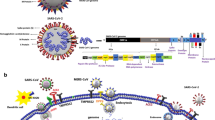
In a review on the central nervous system (CNS) viral infection, Koyuncu et al. concluded that all viruses can reach the CNS under the right conditions depending on viral factors (mutations in specific virulence genes) and host factors (immunodepression, age, and comorbidities) [1]. Respiratory illnesses caused by viral agents, characterized by high rates of morbidity and mortality, are considered problems of critical importance in public health [2,3,4]. Several human respiratory viruses (including coronavirus CoV) are neuroinvasive and neurotropic with potential neuropathological consequences in vulnerable populations. The neurological manifestation seen in patients with viral infection are caused by what is known as the “cytokine storm” including pro-inflammatory and anti-inflammatory cytokines as an immune reaction to the viral infection of the CNS. Such an exaggerated response to the infection can lead to meningitis, encephalitis, meningoencephalitis, or death. The CoVID-19 pandemic, caused by SARS-CoV 2, is a human respiratory virus that causes infection of the respiratory tract and may lead to pneumonia and respiratory failure similar to SARS-CoV, which shows a neuroinvasive and neurotropic capabilities (Fig. 1).

Immune control of viral infections
Neuroinvasion, neuroinflammation, and microglia activation
The central nervous system is a highly protected organ from most viral infections by virtue of external multilayer barriers, the blood-brain barrier, and effective immune responses. However, some viruses can still enter the CNS through the hematogenous or neuronal retrograde routes resulting in debilitating direct immune-mediated pathologies, although invasion of the nervous system has no selective advantage for the host or the pathogen. The virus can infect the PNS or CNS either by direct infection of nerve endings in the tissues and using axonal transport machinery to gain access to the CNS (Figs. 2 and 3) [1], or by infecting cells of the circulatory system that ultimately carry the infection through the blood-brain barrier (BBB) into the CNS. In the hematogenous route, several viruses infect endothelial cells of the BBB or epithelial cells in the choroid plexi and therefore invading neuronal tissue by breaking the blood-cerebrospinal fluid barrier or using leukocytes as a vector for dissemination within the CNS [1]. Once the virus escapes these physical barriers and invades the CNS, the first line of defense is the activation of microglia.

Several viruses spread to the CNS by infecting the neuron receptor in the nasal olfactory epithelium to reach the brain by axonal transport along the olfactory nerve

Some respiratory viruses spread from the lungs to the CNS through the vagus nerve
Microglia are the residing mononuclear phagocytes of the brain, highly heterogeneous cells within the healthy CNS [5], with multiple morphological and functional profiles depending on their environment. They account for about 10% of the total cell population of the brain [6, 7]. The presence of activated glial cells is indicative of neuropathology and considered a marker of brain injury and neuroinflammatory events [8]. However, neuroinflammation has been considered a mediator of secondary damage by secretion of cytokines, neurotrophic factors, and activation of proteases for extracellular matrix remodeling. We now know that molecules of the systemic innate immune system or directly impacted neuronal populations can activate the microglia in the brain and start the neuroinflammatory events. While microglia are not the only cells responsible for the inflammatory or immune-mediated responses in the brain, nevertheless, they can rapidly respond to environmental changes. Recently, there is increasing evidence showing that brain resident glial cells can be transformed into an aggressive effector cells causing neuronal damage [9].
Therefore, microglia may confer short-term neuroprotection or trigger long-term neurodegeneration depending on the interplay between pro- and anti-inflammatory cytokines released in response to viral infection.
Viral infection and neuroinflammation are triggers of neurodegenerative diseases
Viruses can lead to brain dysfunction and neuronal damage by direct cytolytic effects or secondary inflammatory reactions (indirect effect) [10]. Neurotropic viruses have developed mechanisms to escape host immune surveillance to gain access to the CNS. The systemic and local inflammatory responses to viruses are potential causes of neuronal damage. Primary infection of neurons leads to acute cell dysfunction that can result in lethal encephalomyelitis [11]. Activated microglia have been considered to be the main contributors to released cytokine and chemokine. However, recently, there is evidence showing that neuronal cells express specific molecules that can play the role of immune receptors to the immune system to modulating the innate immune response in the brain [12]. These molecules also have a major function in the neuroplasticity and organization of neuronal networks and synapses. Such autonomous activation of neuronal cells using the innate receptors during viral infections could compromise neuroplasticity and the trigger subsequent neuronal dysfunction. Furthermore, viral infection-induced inflammatory events show similarities to those observed in early neurodegenerative conditions, including altered expression of proteins relevant to axonal transport and synaptic transmission ([13]. [14]). In animal models of autoimmune encephalomyelitis (multiple sclerosis), the inflammation targets dendritic spines and leads to synaptic degeneration [15]. Additionally, synaptic dysfunction also occurs typically in the early stages of Alzheimer’s disease pathogenesis [16].
Therefore, inflammatory reactions triggered by viral infections could initiate neurodegeneration, especially in individuals who are already at risk for neurodegenerative disorders as a result of the epigenetic differences that modulate the immune response or the individual’s genetic makeup or susceptibility to infectious diseases [17].
The particularity of respiratory viruses in infected nervous system
Respiratory viruses such as respiratory syncytial virus (RSV) [18, 19], henipaviruses ([20, 21]), influenza A and B ([22]), and enterovirus D68 [23] are also sometimes found in the blood and, being neuroinvasive, they may use the hematogenous route to reach the CNS. These respiratory viruses can also use the olfactory nerve (Fig. 3) to reach the brain [1, 24, 25]. Furthermore, some of these viruses can use other peripheral nerves by targeting nociceptive neuronal cells in the nasal cavity and use the trigeminal nerve [26, 27], or in other cases, the sensory fibers of the vagus nerve in different organs of the respiratory tract [18, 28].
Several viruses spread to the CNS by infecting the neuron receptor in the nasal olfactory epithelium to reach the brain via axonal transport along the olfactory nerve.
Some viruses can spread by infecting the pseudounipolar sensory neurons of the PNS then anterograde axonal transport to CNS.
Influenza Virus (IV) is the most relevant viral etiology agent in respiratory tract infections. Several studies have shown that influenza A can be associated with neurological complications in both children and adults [29,29,31]. Several neurological pathologies related to influenza A infection have been described in the literature, including encephalitis [32], Guillain-Barre syndrome [33, 34], febrile seizure [35], acute necrotizing encephalopathy, and possibly acute disseminated encephalomyelitis (ADEM) [36]. In animal models, studies have shown that the routes of invasion used by IV are either the olfactory route in the nasal cavity or the sensory neurons of the vagus nerve in the lungs (Fig. 4). Thus, influenza A virus can affect cognition and behavior as long-term sequelae by altering the hippocampus and the regulation of neurotransmission [18, 37,38,39,40]. Other studies related to the influenza A virus show the risk of developing neurodegenerative diseases such as Parkinson’s disease (PD) [41].

a Several viruses spread to the CNS by infecting the neuron receptor in the nasal olfactory epithelium to reach the brain by axonal transport along the olfactory nerve. b Some respiratory viruses spread from the lungs to the CNS through the vagus nerve
Coronavirus CoV is a group of non-segmented positive-sense RNA viruses belonging to the Coronaviridae family and the Nidovirales with four subfamilies: AlphaCoV (ACoV), BetaCoV (BCoV), DeltaCoV (DCoV), and GammaCoV [42]. CoV-OC-43 and CoV-229E are described as pathogenic in humans and are responsible for two beta coronaviruses (BCoVs) epidemics: the Middle East respiratory syndrome (MERS-CoV) [43, 44], and Severe Acute Respiratory Syndrome (SARS-CoV) [45, 46].
The WHO declared that in December 2019, a series of viral pneumonia cases of unknown cause has erupted in Wuhan, Hubei, China (WHO Novel coronavirus report January 19, 2020) [47]. A sequencing analysis from respiratory tract samples showed a novel coronavirus, named 2019 novel coronavirus (COVID-19).
The last endemics of SARS-CoV and MERS-CoV showed that CoV could cause extra-pulmonary pathologies most commonly in children as myocarditis, severe diarrhea, and multi-organ failure ([48,49,50,51,52,53]). Several studies described the neurotropic and neuroinvasive capabilities of CoV, through the hematogenous or neuronal retrograde route, leading to neurological pathologies such as multiple sclerosis and encephalomyelitis ([3, 54,55,56]); nevertheless, the capacity of CoV to infect the CNS is not well known ([57, 58]) .
In 1980, Burks was the first to detect CoV in the CNS in an autopsy of patients with MS ([59]). In 2000, Arbour et al. reported a 67% case positive for CoV in a series of autopsy samples from patients with different neurological diseases, especially MS. In 2003, Hung et al. reported the first case of SARS-CoV infection with neurological manifestations [60]; a 59-year-old woman was first admitted with severe respiratory failure and seizures; in addition, the detection was also positive in both tracheal aspirates and CSF samples [60]. Lau et al. reported that viral genetic material was found in CSF samples of a 32-year-old woman with SARS-CoV infection [54]. SARS-CoV organ dissemination was reported in autopsy samples from patients that died of SARS-CoV. The autopsy showed the existence of viral RNA and SARS-CoV-N protein in the small intestine, liver, sweat glands, parathyroid, stomach, kidney, pituitary gland, and cerebrum, confirming the capacity of SARS-CoV to induce a systemic infection [61].
Furthermore, experimental studies show that transgenic mice infected intranasally with SARS-CoV-34 or MERS-CoV-13 had the virus detected in some specific brain areas (thalamus and brainstem) [62]. Some studies detected MERS-CoV virus only in the brain and not in the lungs with a high mortality rate [62]. Interestingly, the brainstem was the most heavily infected area of the brain either by SARS-CoV-34, 35 or MERS-CoV [62].
CoV are respiratory viruses with neurotropic capacities, which allow them to avoid the immune response and cause neurological complications associated with their infection. The mechanisms and routes to reach the CNS have not been well elucidated; the olfactory route and cranial nerves are the most plausible explanation for their invasion of CNS.
SARS-CoV-2 infected nervous system in COVID-19 patients
SARS-CoV and MERS-CoV cause lethal acute pneumonia with similar clinical signs and symptoms reported in pneumonia caused by SARS-CoV-2 in COVID-19 patients [60]. However, the literature suggests that the target of SARS-CoV-2 is the upper airway tract while SARS-CoV targets the lower airway tract, which is the main difference between these two viruses. The common symptoms of COVID-19 reported in a Wuhan local hospitals were fever, dry cough, and respiratory distress, most of which are the characteristic symptoms of COVID-19 ([60, 63, 76]).
As discussed above, the BCoV (SARS-CoV, MERS-CoV, CoV-229E, CoV-OC43) have neuroinvasive capabilities, and neurological manifestations have been documented ([62, 64, 65]). Although the routes of neuroinvasion is not well established, the invasion of peripheral nerve terminals, especially the olfactory and vagus nerve are most likely the reasonable routes that lead to CNS infection ([62, 65]).
Thus far, several studies provide direct evidence for the neurotropism of SARS-CoV-2 [66, 67]. Both SARS-CoV-2 and SARS-CoV bind to the ACE2 receptor to access human cells [68]. Because of the structural similarities and their target receptor, many SARS-CoV-mediated pathologies could also apply to SARS-CoV-2. A multiple number of studies have provided strong evidence for the neurovirulence and neurotropism of SARS-CoV [69]. Several case report studies showed that CSF samples of a SARS patient who presented with tonic–clonic seizures tested positive for SARS-CoV, suggesting possible infection of the CNS by SARS-CoV [54]. Furthermore, Jun et al. isolated SARS-CoV from a specimen of brain tissue of a patient with SARS in which the pathological examination of the brain tissue indicated neuronal necrosis and glial cell hyperplasia [70]. In an autopsy study of eight cases with SARS, the viral particles and their genomic sequence were detected in the brain tissue of all cases. Six of them presented with edema and scattered red degeneration of the neurons [53]. Consistent with this, animal models suggest that the brain was the principal target organ for SARS-CoV in transgenic mice for human ACE2 receptor (hACE2 mice) [71]. These findings of neurovirulence and neuroinvasion in SARS-CoV could provide strong circumstantial evidence of the neurotropic characteristics of SARS-CoV-2.
Similarity between SARS-CoV and SARS-CoV-2
The similarity in the pathologies between SARS-CoV and SARS-CoV-2 is becoming more and more evident. Recent studies showed a 79.5% genomic sequence homology between SARS-CoV and SARS-CoV-2 [131] and genetic similarities to bat coronavirus as high as 96% [72]. In a study based on the Homology and Phylogenetic Analysis of SARS-CoV and SARS-CoV-2 for a comparative purpose, the protein sequence using Blast showed that most of SARS-CoV-2 proteins are highly homologous (95–100%) to the proteins of SARS-CoV virus, in the comparative genomic analyses of SARS-CoV-2 and SARS-CoV using zpicture. The genomic sequences of SARS-CoV-2 and SARS-CoV have a high homology at the nucleotide level, with only six regions of difference (RD) in the genome sequence between SARS-CoV and SARS-CoV-2 [73]. The nucleocapsid protein in SARS-CoV-2 shares ~ 90% amino acid identity with that in SARS-CoV [74]. In addition, spike stalk S2 in SARS-CoV-2 are 99% identical to those of the two bat SARS-like CoVs (bat-SL-CoVZXC21 and bat-SL-CoVZC45) and human SARS-CoV [127].
The neuroinvasive capabilities in SARS-CoV are reported as a common feature of coronaviruses. This leads us to conclude that it is most likely that SARS-CoV-2 has a similar neuropathogenic potential, theoretically. The chronology of SARS-CoV-2 infection process includes manifestation of symptoms after 5 days of infection. Older patients and patients with immunodeficiency may develop respiratory failure in 8 to 15 days [76]. Consistent with this, SARS-CoV-2 has a long enough latency period for the invasion of peripheral nerve terminals which subsequently results in the invasion of the CNS.
Neurological manifestations of SARS-CoV-2 in patients with COVID-19
To date, there are only six reports describing the neurological manifestations of SARS-CoV-2 in patients with COVID-19 (Table 1), in which 173 of 756 patients had neurological manifestation. In a case report of 214 patients with COVID-19 in WHAN, China, neurological manifestation as acute cerebrovascular diseases and impaired consciousness were seen in 36.4% of all patients, and about 88% (78/88) of patients with serious complications ([125]). Moriguchi et al. reported a 24-year-old man admitted to hospital with transient generalized seizures with a Glasgow coma scale of 6 with hemodynamic stability. Before 9 days, the patient felt headaches, generalized fatigue, and fever. The RT-PCR test for SARS-CoV-2 was performed using a nasopharyngeal swab and CSF. Viral nucleic acids were detected in the CSF but not from nasopharyngeal swabs. This patient was diagnosed with meningoencephalitis due to SARS-CoV-2 infection [125].
In Beijing Ditan Hospital, Xiang et al. confirmed the presence of SARS-CoV-2 in the CSF of patients with COVID-19 clinically presented with viral encephalitis [78].
Similarly, Mingxiang et al. reported the same observation in a patient with SARS-CoV-2 presented with a viral meningoencephalitis in which SARS-CoV-2 was detected in the CSF.
SARS-CoV-2 can also damage the spinal cord and lead to acute myelitis [75]. Zahra et al. reports a case of a 65-years-old male admitted with acute progressive symmetric ascending quadriparesis that began with acute progressive weakness of distal lower extremities. The patient had also facial paresis bilaterally 5 days before hospital admission. Two weeks later, the patient suffered from cough, fever, and sometimes dyspnea. RT-PCR for COVID-19 was positive. In this study, Guillain-Barre syndrome in a patient infected with COVID-19 was reported 2020) [128].
Hua et al. reported a case of a 61-years-old woman admitted with progressive weakness in both legs and severe fatigue. Four days before she returned from Wuhan, without fever, cough, chest pain, or diarrhea. The patient was diagnosed with Guillain-Barre syndrome associated with SARS-CoV-2 infection [129].
The olfactory epithelium and olfactory dysfunction in COVID 19 patients
SARS-CoV-2 is characterized by the high infectivity due to glycoprotein spike considered as the important factor accelerating the spread of COVID-19. The spike leads the virus to bind to the host receptor with much higher affinity as compared to related SARS-CoV virus. Recent reports indicate that the early marker of SARS-CoV-2 infection is anosmia or partial loss of the sense of smell (hyposmic). In a study of 357 patients, 85.6% had olfactory dysfunction related to the SARS-CoV-2 infection, 284 (79.6%) patients were anosmic, and 73 (20.4%) were hyposmic [79]. This manifestation may be considered as direct damage to the olfactory receptor neurons located in the olfactory epithelium. Additionally, cells located in the olfactory epithelium (neuronal and nonneuronal cells) express both ACE2 and TMPRSS2 protein receptors required for efficient SARS-CoV-2 infection in humans ([80]. [81]). Some studies showed that TMPRS2 protein receptors is expressed on neuronal cells and likely facilitate SARS-CoV-2 brain infection through the anterograde axonal transport along the olfactory nerve (Fig. 2) [81, 82]. SARS-CoV-2 can reach the brain if the virus first invades high ACE2 expressing, yet unidentified nonneuronal, cells in the olfactory epithelium and then pass to low ACE2 expressing mature neuronal cells to be transported along olfactory axons to the brain.
In animal models, some studies showed that the expression of murine ACE2 and TMPRSS2 have the tendency to increase with age as determined by microarray analysis [82]. If this is the case for humans, then in elderly people, the olfactory epithelium may be more sensitive to SARS-CoV-2 accumulation.
To understand the antiviral protective immune responses of neuronal cells in the olfactory epithelium, a recent study in fish shows that neuronal cells initiate ultrarapid immune responses in the olfactory epithelium after binding rhabdovirus surface glycoprotein [83]. Additionally, the activation of the pro-inflammatory effects inhibits the uptake of the virus into ciliated dendrites/soma and subsequent anterograde axonal transport along the olfactory nerve to reach the brain. As a result, infected neurons undergo apoptosis, which may eliminate initiation of olfactory stimuli and anosmia.
Based on the above observation, infected people with SARS-CoV-2 who show signs of olfactory dysfunctions may actually represent those individuals with faster and stronger initiate immune response against the SARS-CoV-2 infection. Noteworthy that older patients who are much more sensitive to SARS-CoV-2 infection also have degenerative dysfunctions in their neuronal cells in the olfactory epithelium. This is consistent with the observation that elderly patients have more severe COVID-19 symptoms most likely due to the attenuated early immune response.
The anatomical perspective in COVID-19 infection
In experimental studies using transgenic mice, SARS-CoV-34, 35 or MERS-CoV was found in the brain and very heavily in the brainstem [62]. It is known that some respiratory viruses can reach the brain by the retrograde route via infecting the sensory fibers of the vagus nerve in different organs of the respiratory tract [18, 28]. The vagal nucleus consists of four nuclei, including the ambiguous nucleus located in the medulla oblongata in the brainstem [84]. Additionally, the basic respiratory rhythm is generated in the central respiratory neuron network located in various sites in the lower brainstem, more specifically, ventral to the ambiguous nucleus and the dorsal motor nucleus of the vagus, and the area postrema, which are then output as motor activities generated to regulate the respiratory rhythm [85]. Therefore, it is plausible to hypothesize that if SARS-CoV-2 reach the brain through the vagus nerve, the virus will invade the brainstem starting with the vagal nucleus and surrounding sites including the respiratory control center which can lead to more respiratory dysfunction that further exacerbate the damage caused by the primary infection in the lungs.
Neuropathologies and sequelae related to SARS-CoV-2 infections
Some studies describe the persistence of CoV RNA in the human CNS [86]. In experimental studies, mice who survived acute encephalitis caused by CoV showed a long-term sequelae, including the hypo-activity in an open field test, decreased hippocampus excitability with concomitant neuronal loss in the CA1 and CA3[87]. Similar results in studies with infection by the influenza A virus and RSV [88,89,90] infection by West Nile virus (WNV) showed the same neuronal loss in the CA3 [91].
A patient with COVID-19 was diagnosed with necrotizing hemorrhagic encephalitis after presenting with altered mental status. CT images showed a symmetric hypo-attenuation in medial thalami bilaterally and MRI confirmed a hemorrhagic lesions in the bilateral thalami, subinsular regions and medial temporal lobes [92]. In a case report of a 24-year-old man, he presented headaches, generalized fatigue, fever, with generalized seizures, and altered mental status that progressed and led to impaired consciousness. Clinical and laboratory diagnosis was viral meningoencephalitis and RT-PCR analysis of the CSF confirm the SARS-CoV-2 infection. Brain MRI showed changes in the right mesial temporal lobe, the right wall of the lateral ventricle, and the hippocampus. Interestingly, the nasopharyngeal swab specimen for RT-PCR was found to be negative for SARS-CoV-2, which means that COVID-19 may have independent mechanisms of neuropathogenesis [125].
Xu et al. reported in an autopsy study that edema has been detected in the brain tissue of COVID-19 patients due to the hypoxia caused by SARS-CoV-2 [73]. This means COVID-19 patients have the potential to develop infectious toxic encephalopathy due to the hypoxia and viremia. The essential pathological changes in this disease are cerebral edema, with no evidence of inflammation from CSF analysis [130].
A significant amount of evidence shows that respiratory infection is an independent risk factor for acute cerebrovascular disease [94, 95]. Experimental mouse models suggest that some respiratory viruses as influenza can aggravate ischemic brain injury and increase the risk of cerebral hemorrhage by triggering a cytokine storm [96]. As previously discussed, SARS-CoV-2 causes cytokine storm syndromes, which may be one of the factors that patients with COVID-19 can develop acute cerebrovascular disease [63, 97]. Furthermore, SARS-CoV-2 infections often show elevated levels of D-dimer and severe platelet reduction in patients with critical conditions, which may increase the chance of acute cerebrovascular events [76].
Cerebrovascular diseases and SARS-CoV-2 infections
A meta-analysis of 46,248 infected patients in 8 studies from China showed that the most prevalent comorbidities were hypertension (17%) and diabetes mellitus (8%), then cardiovascular diseases (5%) [77, 98]. This indicates that COVID-19 and stroke share similar risk factors. There is strong evidence that the severity of COVID-19 infection is directly linked to the presence of cardiovascular comorbidities [99]. In a case series from Wuhan of 214 patients with COVID-19, 14 patients developed strokes. The study concluded that patients with cardiovascular risk factors were more likely to developed acute cerebrovascular diseases [75]. Similarly, 4 COVID-19 patients with cardiovascular risk developed stroke. The mechanism of vascular damage in all these patients was the large vessel disease [100]. An observational study showed that in 221 COVID-19 patients, 5% developed acute ischemic stroke, 0.5% cerebral hemorrhage, and 0.5% cerebral venous sinus. The incidence was higher in older patients with cardiovascular risk factors [101].
COVID-19 and multiple sclerosis
The etiology of several long-term neuropathologies is still not well known, such as multiple sclerosis (MS), in which a viral infection may play a role in the pathogenesis in genetically predisposed individuals [102,98,99,100,106]. Over the last decades, several viruses have been associated with MS, based on detection of viral nucleic acids, virions, or viral proteins in the CNS or the presence of antiviral antibodies in the serum and/or cerebrospinal fluid. In 1980, for the first time, the association of coronaviruses with MS was suggested for by their isolation from the CNS of two patients [59].
Since then, multiple studies have linked human coronaviruses (including CoV-OC-43 and CoV-229E) with MS. In several autopsy studies, the coronavirus-like particles were detected in brain material obtained from MS patients [107] and in other studies, viruses molecularly related to murine neurotropic coronaviruses were detected in the brain tissue from MS patients [59]. Additionally, anti-HCoV intrathecal antibody synthesis, indicative of a CNS infection, was reported in MS patients [108]. Furthermore, HCoV RNA was detected in the brain [109,110,111] and in the cerebrospinal fluid of MS and other neurological disease (OND) patients [112]. Coronavirus antigens were also detected in MS patient brains [109, 110].
Interestingly, in an autopsy studies of 90 patients with MS, viral RNA was present in a large number of samples; 48% (44 of 90) of all donors were positive for one or both viral strains (CoV-OC-43 and CoV-229E). HCoV-OC43 RNA was widely detected in normal white and gray matter as well as in plaques of MS patient brains. This study also suggested that HCoV-OC43 has the capacity to persistently infect cells in the human CNS and that such infections could lead in some cases to specific molecular adaptation of this virus to the CNS environment [57]. Consistent with this, in animal model studies, a persistent coronavirus infection provoked a chronic demyelination in genetically predisposed mice through immune reaction-mediated mechanisms [113].
There are multiple possible mechanisms by which a viral infection could induce a demyelinating disease such as MS-like lesions. It could be induced as a direct consequence of viral infection such as lytic infection of oligodendrocytes [114]. Alternatively, it could be indirectly mediated through the expression of cytotoxic molecules by glial cells such as MHV-induced chronic demyelination in the CNS. In the latter, several inflammatory molecules, such as IL-6, tumor necrosis factor, interleukin 1β (IL-1β), type 2 nitric oxide synthase [115], RANTES, cytokine response gene 2, and macrophage-inflammatory protein 1β, or their mRNAs, were detected [116].
The HCoV-OC43 virus was detected in the CNS of a child with acute disseminated encephalomyelitis (ADEM) [55] which is another demyelinating neurological disorder characterized by neuroinflammation after nonspecific upper respiratory tract infections. Even though the etiological agent remains unknown, several studies suggest that human coronviruses can lead to ADEM. Subacute panencephalitis sclerosis (SSPE), a progressive fatal neurological disease, is also clearly linked to a viral infection as the persistence of measles virus within the CNS [117].
Viral infection of oligodendrocytes could lead to demyelinating disease through the alteration of their normal function or cytolysis as is the case for reactivated human polyomavirus, which induces the progressive lysis of oligodendrocytes during progressive multifocal leukoencephalopathy [118, 119]. The release of myelin components could also trigger an autoimmune attack. Infection or activation of astrocytes and microglia could lead to release of inflammatory mediators that could damage oligodendrocytes, therefore exacerbating the pathology [120,121,122,123].
It has been shown that CoV-OC-43 is responsible for encephalitis with a T cell response to the infection [124]. This underlines the possibility that the trigger for long-term demyelinating neuropathologies such as MS-like lesions in patients with respiratory viruses is the immune response. Considering the phylogenetic studies and the similarities between SARS-CoV-2 and all beta coronaviruses, including the lineage A groups prototypical coronaviruses, we can conclude that even if the genotype of the virus itself can play a role in the pathogenesis of MS-like lesions associated to viral infection and CoV-OC-43 and CoV-229E may be considered as a risk factor to develop MS. Taking into account the similarities between SARS-CoV-2 and CoV-OC-43 and CoV-229E as well as the characteristics of the immune response to SARS-CoV-2 infection including to the cytokine storm and the surge of IL-6, tumor necrosis factor, interleukin 1β (IL-1β), and type 2 nitric oxide synthase, it is almost certainly that SARS-CoV-2 may be responsible for MS-like demyelinating lesions as a long-term consequences of COVID-19.
Conclusion
Based on the available data, it is highly likely that respiratory distress is not only the result of pulmonary inflammatory structural damage, but also due to the damage caused by the virus in the respiratory centers of the brain, making it more difficult to manage these patients.
Several human respiratory viruses can have neuroinvasive and neurotropic capabilities, leading to neuropathological consequences in vulnerable populations. Understanding the consequences of neuroinvasion and the underlining mechanisms of respiratory viruses (including coronaviruses) and their interactions with the central nervous system is essential as it can be used to better understand the potential pathological relevance of the infection, in addition to the design of novel diagnosis and intervention strategies that will help uncover the potential draggability of molecular virus-host interfaces highly relevant to symptoms of various neurological diseases with a viral involvement.
References
Koyuncu OO, Hogue IB, Enquist LW (2013) Virus infections in the nervous system. Cell Host Microbe 13:379–393. https://doi.org/10.1016/j.chom.2013.03.010
Englund J, Feuchtinger T, Ljungman P (2011) Viral infections in immunocompromised patients. Biol Blood Marrow Transplant 17:S2–S5. https://doi.org/10.1016/j.bbmt.2010.11.008
Talbot HK, Falsey AR (2010) The diagnosis of viral respiratory disease in older adults. Clin Infect Dis 50:747–751. https://doi.org/10.1086/650486
Tregoning JS, Schwarze J (2010) Respiratory viral infections in infants: causes, clinical symptoms, virology, and immunology. Clin Microbiol Rev 23:74–98. https://doi.org/10.1128/cmr.00032-09
Saitgareeva AR, Bulygin KV, Gareev IF, Beylerli OA, Akhmadeeva LR (2020) The role of microglia in the development of neurodegeneration. Neurol Sci. https://doi.org/10.1007/s10072-020-04468-5
Streit WJ (2006) Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci 29:506–510
Streit WJ, Mrak RE, Griffin WS (2004) Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation 1:14
von Bernhardi R (2007) Glial cell dysregulation: a new perspective on Alzheimer Disease. Neurotox Res 12:215–232
Nakajima K, Tohyama Y, Kohsaka S, Kurihara T (2001) Ability of rat microglia to uptake extracellular glutamate. Neurosci Lett 307:171–174
van den Pol AN (2006) Viral infections in the developing and mature brain. Trends Neurosci 29:398–406
Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA (2004) Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 10:1366–1373
Boulanger LM (2009) Immune proteins in brain development and synaptic plasticity. Neuron. 64:93–109
Deleidi M, Hallett PJ, Koprich JB, Chung CY, Isacson O (2010) The Toll-like receptor-3 agonist polyinosinic:polycytidylic acid triggers nigrostriatal dopaminergic degeneration. J Neurosci 30:16091–16101
Chung CY, Koprich JB, Siddiqi H, Isacson O (2009) Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci 29:3365–3373
Centonze D, Muzio L, Rossi S, Cavasinni F, De Chiara V, Bergami A, Musella A, D’Amelio M, Cavallucci V, Martorana A, Bergamaschi A, Cencioni MT, Diamantini A, Butti E, Comi G, Bernardi G, Cecconi F, Battistini L, Furlan R, Martino G (2009) Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J Neurosci 29:3442–3452
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580
Arkwright PD, Abinun M (2008) Recently identified factors predisposing children to infectious diseases. Curr Opin Infect Dis 21:217–222
Bohmwald K, Espinoza JA, Gonzalez PA, Bueno SM, Riedel CA, Kalergis AM (2014) Central nervous system alterations caused by infection with the human respiratory syncytial virus. Rev Med Virol 24:407–419
Halfhide CP, Flanagan BF, Brearey SP, Hunt JA, Fonceca AM, McNamara PS, Howarth D, Edwards S, Smyth RL (2011) Respiratory syncytial virus binds and undergoes transcription in neutrophils from the blood and airways of infants with severe bronchiolitis. J Infect Dis 204:451–458
Mathieu C, Pohl C, Szecsi J, Trajkovic-Bodennec S, Devergnas S, Raoul H, Cosset FL, Gerlier D, Wild TF, Horvat B (2011) Nipah virus usesleukocytes for efficient dissemination within a host. J Virol 85:7863–7871
Escaffre O, Borisevich V, Carmical JR, Prusak D, Prescott J, Feldmann H, Rockx B (2013) Henipavirus pathogenesis in human respiratory epithelial cells. J Virol 87:3284–3294
Tse H, To KK, Wen X, Chen H, Chan KH, Tsoi HW, Li IW, Yuen KY (2011) Clinical and virological factors associated with viremia in pandemic influenza A/H1N1/2009 virus infection. PLoS One 6:e22534
Imamura T, Suzuki A, Lupisan S, Kamigaki T, Okamoto M, Roy CN, Olveda R, Oshitani H (2014) Detection of enterovirus 68 in serum from pediatric patients with pneumonia and their clinical outcomes. Influenza Other Respir Viruses 8:21–24
Berth SH, Leopold PL, Morfini GN (2009) Virus-induced neuronal dysfunction and degeneration. Front Biosci 14:5239–5259
Mori I (2015) Transolfactory neuroinvasion by viruses threatens the human brain. Acta Virol 59:338–349
Lochhead JJ, Thorne RG (2012) Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev 64:614–628
Lochhead JJ, Kellohen KL, Ronaldson PT, Davis TP (2019) Distribution of insulin in trigeminal nerve and brain after intranasal administration. Sci Rep 9:2621
Driessen AK, Farrell MJ, Mazzone SB, McGovern AE (2016) Multiple neural circuits mediating airway sensations: recent advances in the neurobiology of the urge-to-cough. Respir Physiol Neurobiol 226:115–120
Goenka A, Michael BD, Ledger E, Hart IJ, Absoud M, Chow G, Lilleker J, Lunn M, McKee D, Peake D, Pysden K, Roberts M, Carrol ED, Lim M, Avula S, Solomon T, Kneen R (2014) Neurological manifestations of influenza infection in children and adults: results of a national british surveillance study. Clin Infect Dis 58:775–784. https://doi.org/10.1093/cid/cit922
Paksu MS, Aslan K, Kendirli T, Akyildiz BN, Yener N, Yildizdas RD, Davutoglu M, Yaman A, Isikay S, Sensoy G, Tasdemir HA (2018) Neuroinfluenza: evaluation of seasonal influenza associated severe neurological complications in children (a multicenter study). Childs Nerv Syst 34:335–347. https://doi.org/10.1007/s00381-017-3554-3
Popescu CP, Florescu SA, Lupulescu E, Zaharia M, Tardei G, Lazar M, Ceausu E, Ruta SM (2017) Neurologic complications of influenza B virus infection in adults, Romania. Emerg Infect Dis 23:574–581. https://doi.org/10.3201/eid2304.161317
Newland JG, Romero JR, Varman M, Drake C, Holst A, Safranek T, Subbarao K (2003) Encephalitis associated with influenza B virus infection in 2 children and a review of the literature. Clin Infect Dis 36:e87–e95. https://doi.org/10.1086/368184
Ottaviani D, Boso F, Tranquillini E, Gapeni I, Pedrotti G, Cozzio S, Guarrera GM, Giometto B (2020) Early Guillain-Barré syndrome in coronavirus disease 2019 (COVID-19): a case report from an Italian COVID-hospital. Neurol Sci 41:1351–1354. https://doi.org/10.1007/s10072-020-04449-8
Sivadon-Tardy V, Orlikowski D, Porcher R, Sharshar T, Durand MC, Enouf V, Rozenberg F, Caudie C, Annane D, van der Werf S, Lebon P, Raphaël JC, Gaillard JL, Gault E (2009) Guillain-Barré syndrome and influenza virus infection. Clin Infect Dis 48(1):48–56. https://doi.org/10.1086/594124
Chiu SS, Tse CYC, Lau YL, Peiris M (2001) Influenza a infection is an important cause of febrile seizures. Pediatrics 108:E63. https://doi.org/10.1542/peds.108.4.e63
Zeng H, Quinet S, Huang W, Gan Y, Han C, He Y, Wang Y (2013) Clinical and MRI features of neurological complications after influenza A (H1N1) infection in critically ill children. Pediatr Radiol 43:1182–1189
Beraki S, Aronsson F, Karlsson H, Ogren SO, Kristensson K (2005) Influenza a virus infection causes alterations in expression of synaptic regulatory genes combined with changes in cognitive and emotional behaviors in mice. Mol Psychiatry 10:299–308
Hosseini S, Wilk E, Michaelsen-Preusse K, Gerhauser I, Baumgartner W, Geffers R, Schughart K, Korte M (2018) Long-term neuroinflammation induced by influenza a virus infection and the impact on hippocampal neuron morphology and function. J Neurosci 38:3060–3080
Matsuda K, Park CH, Sunden Y, Kimura T, Ochiai K, Kida H, Umemura T (2004) The vagus nerve is one route of transneural invasion for intranasally inoculated influenza a virus in mice. Vet Pathol 41:101–107
Shinya K, Makino A, Hatta M, Watanabe S, Kim JH, Hatta Y, Gao P, Ozawa M, Le QM, Kawaoka Y (2011) Subclinical brain injury caused by H5N1 influenza virus infection. J Virol 85:5202–5207
Jang H, Boltz D, Sturm-Ramirez K, Shepherd KR, Jiao Y, Webster R, Smeyne RJ (2009) Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A 106:14063–14068
King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (Eds) (2012) Classification and nomenclature of viruses. In: Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier: San Diego, 1326–1327
Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc J, Bellini WJ, Anderson LJ, SARS Working Group (2003) A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348:1953–1966
Sheahan TP, Sims AC, Leist SR, Schäfer A, Won J, Brown AJ, Montgomery SA, Hogg A, Babusis D, Clarke MO, Spahn JE, Bauer L, Sellers S, Porter D, Feng JY, Cihlar T, Jordan R, Denison MR, Baric RS (2020) Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat Commun 11:222
de Groot RJ, Baker SC, Baric RS et al (2013) Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J Virol 87:7790–7792
Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820
WHO novel coronavirus – China. http://www.who.int/csr/don/12-january-2020-novel-coronavirus-china/en/ Date: Jan 12, 2020 Date accessed: January 19, 2020
Gerna G, Passarani N, Battaglia M, Rondanelli EG (1985) Human enteric coronaviruses: antigenic relatedness to human coronavirus OC43 and possible etiologic role in viral gastroenteritis. J Infect Dis 151:796–803
Jevsnik M, Steyer A, Pokorn M, Mrvic T, Grosek S, Strle F, Lusa L, Petrovec M (2016) The role of human coronaviruses in children hospitalized for acute bronchiolitis, acute gastroenteritis, and febrile seizures: a 2-year prospective study. PLoS One 11:e0155555
Raj VS, Osterhaus AD, Fouchier RA, Haagmans BL (2014) MERS: emergence of a novel human coronavirus. Curr Opin Virol 5:58–62
Resta S, Luby JP, Rosenfeld CR, Siegel JD (1985) Isolation and propagation of a human enteric coronavirus. Science 229:978–981
Riski H, Hovi T (1980) Coronavirus infections of man associated with diseases other than the common cold. J Med Virol 6:259–265
Jiang G, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, Zou W, Zhan J, Wang S, Xie Z, Zhuang H, Wu B, Zhong H, Shao H, Fang W, Gao D, Pei F, Li X, He Z, Xu D, Shi X, Anderson VM, Leong AS-Y (2005) Multiple organ infection and the pathogenesis of SARS. J Exp Med 202(3):415–424
Lau KK, Yu WC, Chu CM, Lau ST, Sheng B, Yuen KY (2004) Possible central nervous system infection by SARS coronavirus. Emerg Infect Dis 10(2):342–344. https://doi.org/10.3201/eid1002.030638
Yeh EA, Collins A, Cohen ME, Duffner PK, Faden H (2004) Detection of coronavirus in the central nervous system of a child with acute disseminated encephalomyelitis. Pediatrics 113:e73–e76. https://doi.org/10.1542/peds.113.1.e73
Zlateva KT, Van Ranst M (2004) Detection of subgroup B respiratory syncytial virus in the cerebrospinal fluid of a patient with respiratory syncytial virus pneumonia. Pediatr Infect Dis J 23:1065–1066. https://doi.org/10.1097/01.inf.0000143654.12493.c9
Arbour N, Day R, Newcombe J, Talbot PJ (2000) Neuroinvasion by human respiratory coronaviruses. J Virol 74:8913–8921. https://doi.org/10.1128/jvi.74.19.8913-8921.2000
Desforges M, Le Coupanec A, Brison É, Meessen-Pinard M, Talbot PJ (2014) Neuroinvasive and neurotropic human respiratory coronaviruses: potential neurovirulent agents in humans. In: Adhikari R, Thapa S (eds) Infectious Diseases and Nanomedicine I. Springer, India, pp 75–96
Burks JS, DeVald BL, Jankovsky LD, Gerdes JC (1980) Two coronaviruses isolated from central nervous system tissue of two multiple sclerosis patients. Science 209:933–934. https://doi.org/10.1126/science.7403860
Hung ECW, Chim SSC, Chan PKS, Tong YK, Ng EKO, Chiu RWK, Leung CB, Sung JJY, Tam JS, Lo YMD (2003) Detection of SARS coronavirus RNA in the cerebrospinal fluid of a patient with severe acute respiratory syndrome. Clin Chem 49:2108–2109. https://doi.org/10.1373/clinchem.2003.025437
Ding YQ, He L, Zhang QL, Huang ZX, Che XY, Hou JL et al (2004) Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J Pathol 203:622–630. https://doi.org/10.1002/path.1560
Li Z, He W, Lan Y, Zhao K, Lv X, Lu H, Ding N, Zhang J, Shi J, Shan C, Gao F (2016) The evidence of porcine hemagglutinating encephalomyelitis virus induced nonsuppurative encephalitis as the cause of death in piglets. Peer J 4:e2443
Chen C, Zhang XR, Ju ZY, He WF (2020) Advances in the research of cytokine storm mechanism induced by Corona Virus Disease 2019 and the corresponding immunotherapies. Zhonghua Shao Shang Za Zhi 36:E005
Mengeling WL, Boothe AD, Ritchie AE (1972) Characteristics of a coronavirus (strain 67N) of pigs. Am J Vet Res 33(2):297–308
Andries K, Pensaert MB (1980) Virus isolated and immunofluorescence in different organs of pigs infected with hemagglutinating encephalomyelitis virus. Am J Vet Res 41:215–218
Gialluisi A, de Gaetano G, Iacoviello L (2020) New challenges from Covid-19 pandemic: an unexpected opportunity to enlighten the link between viral infections and brain disorders? Neurol Sci 41:1349–1350. https://doi.org/10.1007/s10072-020-04444-z
Niazkar HR, Zibaee B, Nasimi A, Bahri N (2020) The neurological manifestations of COVID-19: a review article. Neurol Sci. https://doi.org/10.1007/s10072-020-04486-3
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, Tan W (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 395(10224):565–574. https://doi.org/10.1016/S0140-6736(20)30251-8
Tsai LK, Hsieh ST, Chang YC (2005) Neurological manifestations in severe acute respiratory syndrome. Acta Neurol Taiwanica 14(3):113–119
Xu J, Zhong S, Liu J, Li L, Li Y, Wu X, Li Z, Deng P, Zhang J, Zhong N, Ding Y, Jiang Y (2005) Detection of severe acute respiratory syndrome coronavirus in the brain: potential role of the chemokine mig in pathogenesis. Clin Infect Dis 41(8):1089–1096. https://doi.org/10.1086/444461
McCray PB Jr, Pewe L, Wohlford-Lenane C, Hickey M, Manzel L, Shi L, Netland J, Jia HP, Halabi C, Sigmund CD, Meyerholz DK, Kirby P, Look DC, Perlman S (2007) Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol 81(2):813–821. https://doi.org/10.1128/JVI.02012-06
Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, Meng J, Zhu Z, Zhang Z, Wang J, Sheng J, Quan L, Xia Z, Tan W, Cheng G, Jiang T (2020) Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 27(3):325–328
Xu J, Zhao S, Teng T et al (2020) Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 12(2):244. Published 2020 Feb 22. https://doi.org/10.3390/v12020244
Gralinski LE, Menachery VD (2020) Return of the coronavirus: 2019-nCoV. Viruses. 12:135. https://doi.org/10.3390/v12020135
Mao L, Jin H, Wang M, et al (2020) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 77(6):683–690. https://doi.org/10.1001/jamaneurol.2020.1127
Wang W, Xu Y, Gao R, Lu R, Han K, Wu G, Tan W (2020) Detection of SARS-CoV-2 in different types of clinical specimens. JAMA. 323(18):1843–1844. https://doi.org/10.1001/jama.2020.3786
Yang J (2020) Prevalence of comorbidities in the novel Wuhan coronavirus (COVID-19) infection: a systematic review and meta-analysis. Int J Infect Dis 94:91–95
Xiang P, Xu XM, Gao LL, Wang HZ, Xiong HF, Li RH et al. First case of 2019 novel coronavirus disease with encephalitis ChinaXiv, T202003 (2020), p. 00015
Altwairqi RG, Aljuaid SM, Alqahtani AS (2020) Effect of tonsillectomy on humeral and cellular immunity: a systematic review of published studies from 2009 to 2019. Eur Arch Otorhinolaryngol 277(1):1–7. https://doi.org/10.1007/s00405-019-05672-6
Olender T, Keydar I, Pinto JM, Tatarskyy P, Alkelai A, Chien M-S, Fishilevich S, Restrepo D, Matsunami H, Gilad Y, Lancet D (2016) The human olfactory transcriptome. BMC Genomics 17:619
Kangeswaran N, Demond M, Nagel M (2015) Deep sequencing of the murine olfactory receptor transcriptome. PLoS One 10(1):e0113170
Saraiva LR, Ibarra-Soria X, Khan M, Omura M, Scialdone A, Mombaerts P, Marioni JC, Logan DW (2015) Hierarchical deconstruction of mouse olfactory sensory neurons: from whole mucosa to single-cell RNA-seq. Sci Rep 5:18178
Sepahi A, Kraus A, Casadei E, Johnston CA, Galindo-Villegas J, Kelly C, Garcia-Moreno D, Munoz P, Mulero V, Huertas M, Salinas I (2019) Olfactory sensory neurons mediate ultrarapid antiviral immune responses in a TrkA-dependent manner. Proc Natl Acad Sci U S A 116(25):12428–12436
Baker E, Lui F (2020) Neuroanatomy, vagal nerve nuclei (Nucleus Vagus). In: StatPearls. StatPearls Publishing, Treasure Island (FL)
Ikeda K, Kawakami K, Onimaru H, Okada Y, Yokota S, Koshiya N, Oku Y, Iizuka M, Koizumi H (2017) The respiratory control mechanisms in the brainstem and spinal cord: integrative views of the neuroanatomy and neurophysiology. J Physiol Sci 67(1):45–62. https://doi.org/10.1007/s12576-016-0475-y
Arbour N, Cote G, Lachance C, Tardieu M, Cashman NR, Talbot PJ (1999) Acute and persistent infection of human neural cell lines by human coronavirus OC43. J Virol 73:3338–3350
Jacomy H, Fragoso G, Almazan G, Mushynski WE, Talbot PJ (2006) Human coronavirus OC43 infection induces chronic encephalitis leading to disabilities in BALB/C mice. Virology 349:335–346
Espinoza JA, Bohmwald K, Cespedes PF, Gomez RS, Riquelme SA, Cortes CM, Valenzuela JA, Sandoval RA, Pancetti FC, Bueno SM, Riedel CA, Kalergis AM (2013a) Impaired learning resulting from respiratory syncytial virus infection. Proc Natl Acad Sci U S A 110:9112–9117
Espinoza JA, Bohmwald K, Cespedes PF, Gomez RS, Riquelme SA, Cortes CM, Valenzuela JA, Sandoval RA, Pancetti FC, Bueno SM, Riedel CA, Kalergis AM (2013b) Impaired learning resulting from respiratory syncytial virusinfection. Proc Natl Acad Sci U S A 110:9112–9117
Jurgens HA, Amancherla K, Johnson RW (2012) Influenza infection induces neuroinflammation, alters hippocampal neuron morphology, and impairs cognition in adult mice. J Neurosci 32:3958–3968
Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Yu J, Perez-Torres C, Frouin A, Wilton DK, Funk K, DeMasters BK, Jiang X, Bowen JR, Mennerick S, Robinson JK, Garbow JR, Tyler KL, Suthar MS, Schmidt RE, Stevens B, Klein RS (2016) A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534:538–543
Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B (2020) COVID-19-associated Acute Hemorrhagic Necrotizing Encephalopathy: CT and MRI Features [published online ahead of print, 2020 Mar 31]. Radiology. 201187. https://doi.org/10.1148/radiol.2020201187
Mizuguchi M, Yamanouchi H, Ichiyama T, Shiomi M (2007) Acute encephalopathy associated with influenza and other viral infections. Acta Neurol Scand Suppl 186:45–56
Elkind MS (2007) Why now? Moving from stroke risk factors to stroke triggers. Curr Opin Neurol 20(1):51–57
Warren-Gash C, Blackburn R, Whitaker H, McMenamin J, Hayward AC (2018) Laboratory-confirmed respiratory infections as triggers for acute myocardial infarction and stroke: a self-controlled case series analysis of national linked datasets from Scotland. Eur Respir J 51(3):1701794. https://doi.org/10.1183/13993003.01794-2017
Muhammad S, Haasbach E, Kotchourko M, Strigli A, Krenz A, Ridder DA et al (2011) Influenza virus infection aggravates stroke outcome. Stroke 42(3):783–791
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ (2020) COVID-19: consider cytokine storm syndromes and immunosuppression [published online ahead of print, 2020 Mar 16]. Lancet S0140-6736(20):30628
Zhou F (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 395(10229):1054–1062
Guo J (2020) Coronavirus disease 2019 (COVID-19) and cardiovascular disease: a viewpoint on the potential influence of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers on onset and severity of severe acute respiratory syndrome coronavirus 2 infection. J Am Heart Assoc 9(7):e016219
Padroni M (2020) Guillain-Barre syndrome following COVID-19: new infection, old complication? J Neurol 267:1877–1879
El Otmani H, El Moutawakil B, Rafai MA et al (2020) Covid-19 and Guillain-Barré syndrome: More than a coincidence!. Rev Neurol (Paris). 176(6):518–519. https://doi.org/10.1016/j.neurol.2020.04.007
Cusick MF, Libbey JE, Fujinami RS (2013) Multiple sclerosis: autoimmunity and viruses. Curr Opin Rheumatol 25:496–501
Gilden DH (2005) Infectious causes of multiple sclerosis. Lancet Neurol 4:195–202
Kurtzke JF (1993) Epidemiologic evidence for multiple sclerosis as an infection. Clin Microbiol Rev 6:382–427
Saberi A, Akhondzadeh S, Kazemi S (2018) Infectious agents and different course of multiple sclerosis: a systematic review. Acta Neurol Belg 118:361–377
Smatti MK, Cyprian FS, Nasrallah GK, Al Thani AA, Almishal RO, Yassine HM (2019) Viruses and autoimmunity: a review on the potential interaction and molecular mechanisms. Viruses 11:762
Tanaka R, Iwasaki Y, Koprowski H (1976) Intracisternal virus-like particles in brain of a multiple sclerosis patient. J Neurol Sci 28(1):121–126
Salmi A, Ziola B, Hovi T, Reunanen M (1982) Antibodies to coronaviruses OC43 and 229E in multiple sclerosis patients. Neurology. 32:292–295
Murray RS, Brown B, Brian D, Cabirac GF (1992a) Detection of coronavirus RNA and antigen in multiple sclerosis brain. Ann Neurol 31:525–533
Murray RS, Brown B, Brian D, Cabirac GF (1992b) Detection of coronavirus RNA and antigen in multiple sclerosis brain. Ann Neurol 31:525–533
Sorensen O, Coulter-Mackie MB, Puchalski S, Dales S (1984) In vivo and in vitro models of demyelinating disease. IX. Progression of JHM virus infection in the central nervous system of the rat during overt and asymptomatic phases. Virology. 137:347–357
Cristallo A, Gambaro F, Biamonti G, Ferrante P, Battaglia M, Cereda PM (1997) Human coronavirus polyadenylated RNA sequences in cerebrospinal fluid from multiple sclerosis patients. Microbiologica. 2:105–114
Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD (2004) Theiler’s virus infection: a model for multiple sclerosis. Clin Microbiol Rev 17(1):174–207. https://doi.org/10.1128/cmr.17.1.174-207.2004
Weber T, Major EO (1997) Progressive multifocal leukoencephalopathy: molecular biology, pathogenesis and clinical impact. Intervirology. 40:98–111
Sun N, Grzybicki D, Castro F, Murphy S, Perlman S (1995) Activation of astrocytes in the spinal cord of mice chronically infected with a neurotropic coronavirus. Virology. 213:482–493
Lane TE, Asensio VC, Yu N, Paoletti AD, Campbell IL, Buchmeier MJ (1998) Dynamic regulation of α- and β-chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J Immunol 160:970–978
Rima BK, Duprex WP (2005) Molecular mechanisms of measles virus persistence. Virus Res 111(2):132–147
Roberts MT (2005) AIDS-associated progressive multifocal leukoencephalopathy: current management strategies. CNS Drugs 19(8):671–682
Sweet TM, Del Valle L, Khalili K (2002) Molecular biology and immunoregulation of human neurotropic JC virus in CNS. J Cell Physiol 191(3):249–256
Gonzalez-Scarano F, Baltuch G (1999) Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci 22:219–240
Hovelmeyer N, Hao Z, Kranidioti K, Kassiotis G, Buch T, Frommer F, von Hoch L, Kramer D, Minichiello L, Kollias G, Lassmann H, Waisman A (2005) Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J Immunol 175(9):5875–5884
McLarnon JG, Michikawa M, Kim SU (1993) Effects of tumor necrosis factor on inward potassium current and cell morphology in cultured human oligodendrocytes. Glia 9(2):120–126
Miller SD, Vanderlugt CL, Begolka WS, Pao W, Neville KL, Yauch RL, Kim BS (1997) Epitope spreading leads to myelin-specific autoimmune responses in SJL mice chronically infected with Theiler’s virus. J Neuro-Oncol 3(Suppl 1):S62–S65
Butler N, Pewe L, Trandem K, Perlman S (2006) Murine encephalitis caused by HCoV-OC43, a human coronavirus with broad species specificity, is partly immune-mediated. Virology 347:410–421
Moriguchi T (2020) A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis 94:55–58
Yu WB, Tang GD, Zhang L, Corlett RT (2020) Decoding the evolution and transmissions of the novel pneumonia coronavirus (SARS-CoV-2) using whole genomic data. ChinaXiv. https://doi.org/10.12074/202002.00033
Chan JF-W, Kok K-H, Zheng Z, Chu H, To KK-W, Yuan S, Yuen K-Y (2020) Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 9(1):221–236. https://doi.org/10.1080/22221751.2020.1719902
Sedaghat Z, Karimi N (2020) Guillain Barre syndrome associated with COVID-19 infection: A case report. J Clin Neurosci 76:233–235
Zhao H, Shen D, Zhou H, Liu J, Chen S (2020) Guillain-Barré syndrome associated with SARS-CoV-2 infection: causality or coincidence? Lancet Neurol 19(5):383–384
Dobbs M (2011) Toxic Encephalopathy. Semin Neurol 31(02):184–193
Yu F, Du L, Ojcius DM, Pan C, Jiang S (2020) Measures for diagnosing and treating infections by a novel coronavirus responsible for a pneumonia outbreak originating in Wuhan, China. Microbes Infect 22:74–79. https://doi.org/10.1016/j.micinf.2020.01.003
Chan JF-W, Yuan S, Kok K-H, To KK-W, Chu H, Yang J, Xing F, Liu J, Yip CC-Y, Poon RW-S, Tsoi H-W, Lo SK-F, Chan K-H, Poon VK-M, Chan W-M, Ip JD, Cai J-P, Cheng VC-C, Chen H, Hui CK-M, Yuen K-Y (2020) A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395(10223):514–523
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395(10223):497–506
Zhang G, Hu C, Luo L, Fang F, Chen Y, Li J et al (2020) Clinical features and short-term outcomes of 221 patients with COVID-19 in Wuhan, China. J Clin Virol 104364
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Ethical approval was waived by the local Ethics Committee of Cadi Ayyad University, Faculty of Medicine and Pharmacy Marrakech in view of the retrospective nature of the study and all the procedures being performed were part of the routine care.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yachou, Y., El Idrissi, A., Belapasov, V. et al. Neuroinvasion, neurotropic, and neuroinflammatory events of SARS-CoV-2: understanding the neurological manifestations in COVID-19 patients. Neurol Sci 41, 2657–2669 (2020). https://doi.org/10.1007/s10072-020-04575-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-020-04575-3