
Abstract
Introduction
Genetic (idiopathic) generalized epilepsy (GGE) is a common form of epilepsy characterized by unknown aetiology and a presence of genetic component in its predisposition.
Methods
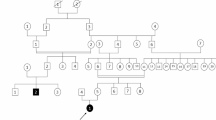
To understand the genetic factor in a family with GGE, we performed whole exome sequencing (WES) on a trio of a juvenile myoclonic epilepsy/febrile seizure (JME/FS) proband with JME/FS mother and healthy father. Sanger sequencing was carried out for validation of WES results and variant detection in other family members.
Results
Predictably damaging variant found in affected proband and mother but absent in healthy father in SCN1A gene was found to be associated with generalized epilepsy and febrile seizure. The novel non-synonymous substitution (c.5753C>T, p.S1918F) in SCN1A was found in all family members with GGE, of which 4/8 were JME subtypes, and/or febrile seizure, while 3 healthy family member controls did not have the mutation. This mutation was also absent in 41 GGE patients and 414 healthy Malaysian Chinese controls.
Conclusion
The mutation is likely to affect interaction between the sodium channel and calmodulin and subsequently interrupt calmodulin-dependent modulation of the channel.




Similar content being viewed by others

References
Lucarini N, Verrotti A, Napolioni V, Bosco G, Curatolo P (2007) Genetic polymorphisms and idiopathic generalized epilepsies. Pediatr Neurol 37(3):157–164. https://doi.org/10.1016/j.pediatrneurol.2007.06.001
Fjaer R, Brodtkorb E, Oye AM, Sheng Y, Vigeland MD, Kvistad KA, Backe PH, Selmer KK (2015) Generalized epilepsy in a family with basal ganglia calcifications and mutations in SLC20A2 and CHRNB2. Eur J Med Genet 58(11):624–628. https://doi.org/10.1016/j.ejmg.2015.10.005
Liu XW, Li W, Han T, Wei K, Qiao S, Su L, Chi Z (2018) The finding of a new heterozygous mutation site of the SCN2A gene in a monozygotic twin family carrying and exhibiting genetic epilepsy with febrile seizures plus (GEFS+) using targeted next-generation sequencing. Clin Neurol Neurosurg 169:86–91. https://doi.org/10.1016/j.clineuro.2017.10.020
Parihar R, Ganesh S (2013) The SCN1A gene variants and epileptic encephalopathies. J Hum Genet 58(9):573–580. https://doi.org/10.1038/jhg.2013.77
Binini N, Sancini G, Villa C, Dal Magro R, Sansoni V, Rusconi R, Mantegazza M, Grioni D, Talpo F, Toselli M, Combi R (2017) Identification of two mutations in cis in the SCN1A gene in a family showing genetic epilepsy with febrile seizures plus (GEFS+) and idiopathic generalized epilepsy (IGE). Brain Res 1677:26–32. https://doi.org/10.1016/j.brainres.2017.09.023
Thounaojam R, Langbang L, Itisham K, Sobhani R, Srivastava S, Ramanujam B, Verma R, Tripathi M, Aguan K (2017) EFHC1 mutation in Indian juvenile myoclonic epilepsy patient. Epilepsia Open 2(1):84–89. https://doi.org/10.1002/epi4.12037
Phani NM, Acharya S, Xavy S, Bhaskaranand N, Bhat MK, Jain A, Rai PS, Satyamoorthy K (2014) Genetic association of KCNJ10 rs1130183 with seizure susceptibility and computational analysis of deleterious non-synonymous SNPs of KCNJ10 gene. Gene 536(2):247–253. https://doi.org/10.1016/j.gene.2013.12.026
Kahle KT, Merner ND, Friedel P, Silayeva L, Liang B, Khanna A, Shang Y, Lachance-Touchette P, Bourassa C, Levert A, Dion PA, Walcott B, Spiegelman D, Dionne-Laporte A, Hodgkinson A, Awadalla P, Nikbakht H, Majewski J, Cossette P, Deeb TZ, Moss SJ, Medina I, Rouleau GA (2014) Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep 15(7):766–774. https://doi.org/10.15252/embr.201438840
Striano P, Weber YG, Toliat MR, Schubert J, Leu C, Chaimana R, Baulac S, Guerrero R, LeGuern E, Lehesjoki AE, Polvi A, Robbiano A, Serratosa JM, Guerrini R, Nurnberg P, Sander T, Zara F, Lerche H, Marini C, Consortium E (2012) GLUT1 mutations are a rare cause of familial idiopathic generalized epilepsy. Neurology 78(8):557–562. https://doi.org/10.1212/WNL.0b013e318247ff54
Jiang Y, Zhang Y, Zhang P, Sang T, Zhang F, Ji T, Huang Q, Xie H, Du R, Cai B, Zhao H, Wang J, Wu Y, Wu H, Xu K, Liu X, Chan P, Wu X (2012) NIPA2 located in 15q11.2 is mutated in patients with childhood absence epilepsy. Hum Genet 131(7):1217–1224. https://doi.org/10.1007/s00439-012-1149-3
Neubauer BA, Waldegger S, Heinzinger J, Hahn A, Kurlemann G, Fiedler B, Eberhard F, Muhle H, Stephani U, Garkisch S, Eeg-Olofsson O, Muller U, Sander T (2008) KCNQ2 and KCNQ3 mutations contribute to different idiopathic epilepsy syndromes. Neurology 71(3):177–183. https://doi.org/10.1212/01.wnl.0000317090.92185.ec
Lachance-Touchette P, Martin C, Poulin C, Gravel M, Carmant L, Cossette P (2010) Screening of GABRB3 in French-Canadian families with idiopathic generalized epilepsy. Epilepsia 51(9):1894–1897. https://doi.org/10.1111/j.1528-1167.2010.02642.x
Saint-Martin C, Gauvain G, Teodorescu G, Gourfinkel-An I, Fedirko E, Weber YG, Maljevic S, Ernst JP, Garcia-Olivares J, Fahlke C, Nabbout R, LeGuern E, Lerche H, Poncer JC, Depienne C (2009) Two novel CLCN2 mutations accelerating chloride channel deactivation are associated with idiopathic generalized epilepsy. Hum Mutat 30(3):397–405. https://doi.org/10.1002/humu.20876
Singh B, Monteil A, Bidaud I, Sugimoto Y, Suzuki T, Hamano S, Oguni H, Osawa M, Alonso ME, Delgado-Escueta AV, Inoue Y, Yasui-Furukori N, Kaneko S, Lory P, Yamakawa K (2007) Mutational analysis of CACNA1G in idiopathic generalized epilepsy. Mutation in brief #962. Online. Hum Mutat 28(5):524–525. https://doi.org/10.1002/humu.9491
Marini C, Porro A, Rastetter A, Dalle C, Rivolta I, Bauer D, Oegema R, Nava C, Parrini E, Mei D, Mercer C, Dhamija R, Chambers C, Coubes C, Thevenon J, Kuentz P, Julia S, Pasquier L, Dubourg C, Carre W, Rosati A, Melani F, Pisano T, Giardino M, Innes AM, Alembik Y, Scheidecker S, Santos M, Figueiroa S, Garrido C, Fusco C, Frattini D, Spagnoli C, Binda A, Granata T, Ragona F, Freri E, Franceschetti S, Canafoglia L, Castellotti B, Gellera C, Milanesi R, Mancardi MM, Clark DR, Kok F, Helbig KL, Ichikawa S, Sadler L, Neupauerova J, Lassuthova P, Sterbova K, Laridon A, Brilstra E, Koeleman B, Lemke JR, Zara F, Striano P, Soblet J, Smits G, Deconinck N, Barbuti A, DiFrancesco D, LeGuern E, Guerrini R, Santoro B, Hamacher K, Thiel G, Moroni A, DiFrancesco JC, Depienne C (2018) HCN1 mutation spectrum: from neonatal epileptic encephalopathy to benign generalized epilepsy and beyond. Brain 141(11):3160–3178. https://doi.org/10.1093/brain/awy263
Epi25 Collaborative. Electronic address sbuea, Epi C (2019) Ultra-rare genetic variation in the epilepsies: a whole-exome sequencing study of 17,606 individuals. Am J Hum Genet 105(2):267–282. https://doi.org/10.1016/j.ajhg.2019.05.020
Yang C, Hua Y, Zhang W, Xu J, Xu L, Gao F, Jiang P (2018) Variable epilepsy phenotypes associated with heterozygous mutation in the SCN9A gene: report of two cases. Neurol Sci 39(6):1113–1115. https://doi.org/10.1007/s10072-018-3300-y
Heinzen EL, Depondt C, Cavalleri GL, Ruzzo EK, Walley NM, Need AC, Ge D, He M, Cirulli ET, Zhao Q, Cronin KD, Gumbs CE, Campbell CR, Hong LK, Maia JM, Shianna KV, McCormack M, Radtke RA, O’Conner GD, Mikati MA, Gallentine WB, Husain AM, Sinha SR, Chinthapalli K, Puranam RS, McNamara JO, Ottman R, Sisodiya SM, Delanty N, Goldstein DB (2012) Exome sequencing followed by large-scale genotyping fails to identify single rare variants of large effect in idiopathic generalized epilepsy. Am J Hum Genet 91(2):293–302. https://doi.org/10.1016/j.ajhg.2012.06.016
Chen ZH, Wang C, Zhuo MQ, Zhai QX, Chen Q, Guo YX, Zhang YX, Gui J, Tang ZH, Zeng XL (2017) Exome sequencing identified a novel missense mutation c.464G>A (p.G155D) in Ca(2+)-binding protein 4 (CABP4) in a Chinese pedigree with autosomal dominant nocturnal frontal lobe epilepsy. Oncotarget 8(45):78940–78947. https://doi.org/10.18632/oncotarget.20694
Carvill GL, Weckhuysen S, McMahon JM, Hartmann C, Moller RS, Hjalgrim H, Cook J, Geraghty E, O'Roak BJ, Petrou S, Clarke A, Gill D, Sadleir LG, Muhle H, von Spiczak S, Nikanorova M, Hodgson BL, Gazina EV, Suls A, Shendure J, Dibbens LM, De Jonghe P, Helbig I, Berkovic SF, Scheffer IE, Mefford HC (2014) GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology 82(14):1245–1253. https://doi.org/10.1212/WNL.0000000000000291
Zeng Q, Yang X, Zhang J, Liu A, Yang Z, Liu X, Wu Y, Wu X, Wei L, Zhang Y (2018) Genetic analysis of benign familial epilepsies in the first year of life in a Chinese cohort. J Hum Genet 63(1):9–18. https://doi.org/10.1038/s10038-017-0359-x
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–2079. https://doi.org/10.1093/bioinformatics/btp352
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498. https://doi.org/10.1038/ng.806
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47(D1):D886–D894. https://doi.org/10.1093/nar/gky1016
Quang D, Chen Y, Xie X (2015) DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31(5):761–763. https://doi.org/10.1093/bioinformatics/btu703
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4(7):1073–1081. https://doi.org/10.1038/nprot.2009.86
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249. https://doi.org/10.1038/nmeth0410-248
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the functional effect of amino acid substitutions and indels. PLoS One 7(10):e46688. https://doi.org/10.1371/journal.pone.0046688
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Capriotti E, Fariselli P, Casadio R (2005) I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res 33(Web Server issue):W306–W310. https://doi.org/10.1093/nar/gki375
Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372(3):774–797. https://doi.org/10.1016/j.jmb.2007.05.022
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O'Donnell-Luria AH, Vallabh Minikel E, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, , Neale BM, Daly MJ, MacArthur DG (2019) Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv
Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR (2015) A global reference for human genetic variation. Nature 526(7571):68–74. https://doi.org/10.1038/nature15393
Bahler M, Rhoads A (2002) Calmodulin signaling via the IQ motif. FEBS Lett 513(1):107–113
Tuncer GO, Teber S, Albayrak P, Kutluk MG, Deda G (2018) A case of Dravet Syndrome with a newly defined mutation in the SCN1A gene. Turk Pediatri Ars 53(4):259–262. https://doi.org/10.5152/TurkPediatriArs.2018.4197
Mahoney K, Moore SJ, Buckley D, Alam M, Parfrey P, Penney S, Merner N, Hodgkinson K, Young TL (2009) Variable neurologic phenotype in a GEFS+ family with a novel mutation in SCN1A. Seizure 18(7):492–497. https://doi.org/10.1016/j.seizure.2009.04.009
Hoffman-Zacharska D, Szczepanik E, Terczynska I, Goszczanska-Ciuchta A, Zalewska-Miszkurka Z, Tataj R, Bal J (2015) From focal epilepsy to Dravet syndrome--Heterogeneity of the phenotype due to SCN1A mutations of the p.Arg1596 amino acid residue in the Nav1.1 subunit. Neurol Neurochir Pol 49(4):258–266. https://doi.org/10.1016/j.pjnns.2015.06.006
Salgueiro-Pereira AR, Duprat F, Pousinha PA, Loucif A, Douchamps V, Regondi C, Ayrault M, Eugie M, Stunault MI, Escayg A, Goutagny R, Gnatkovsky V, Frassoni C, Marie H, Bethus I, Mantegazza M (2019) A two-hit story: Seizures and genetic mutation interaction sets phenotype severity in SCN1A epilepsies. Neurobiol Dis 125:31–44. https://doi.org/10.1016/j.nbd.2019.01.006
Jingami N, Matsumoto R, Ito H, Ishii A, Ihara Y, Hirose S, Ikeda A, Takahashi R (2014) A novel SCN1A mutation in a cytoplasmic loop in intractable juvenile myoclonic epilepsy without febrile seizures. Epileptic Disord 16(2):227–231. https://doi.org/10.1684/epd.2014.0657
Escayg A, Goldin AL (2010) Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51(9):1650–1658. https://doi.org/10.1111/j.1528-1167.2010.02640.x
Catterall WA (2000) From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26(1):13–25
Spampanato J, Kearney JA, de Haan G, McEwen DP, Escayg A, Aradi I, MacDonald BT, Levin SI, Soltesz I, Benna P, Montalenti E, Isom LL, Goldin AL, Meisler MH (2004) A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for beta subunit interaction. J Neurosci 24(44):10022–10034. https://doi.org/10.1523/JNEUROSCI.2034-04.2004
Rusconi R, Combi R, Cestele S, Grioni D, Franceschetti S, Dalpra L, Mantegazza M (2009) A rescuable folding defective Nav1.1 (SCN1A) sodium channel mutant causes GEFS+: common mechanism in Nav1.1 related epilepsies? Hum Mutat 30(7):E747–E760. https://doi.org/10.1002/humu.21041
Acknowledgments
We are grateful to the affected family and all individuals participating in this study.
Funding
This research is supported by High Impact Research MOHE Grant (UM.C/625/1/HIR/MOHE/MED/45) from the Ministry of Higher Education Malaysia.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Informed consent
This study was approved by the Medical Ethics Committee of University Malaya Medical Centre (MEC reference number: 944.3). Written consent was obtained from all subjects.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chan, CK., Low, J.SY., Lim, KS. et al. Whole exome sequencing identifies a novel SCN1A mutation in genetic (idiopathic) generalized epilepsy and juvenile myoclonic epilepsy subtypes. Neurol Sci 41, 591–598 (2020). https://doi.org/10.1007/s10072-019-04122-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-019-04122-9