
Abstract
Advanced biophysical imaging techniques, such as cryo-electron microscopy or tomography, enable 3D volumetric reconstructions of large macromolecular complexes in a near-native environment. However, pure volumetric data is insufficient for a detailed understanding of the underlying protein–protein interactions. This obstacle can be overcome by assembling an atomic model of the whole protein complex from known atomic structures, which are available from either X-ray crystallography or homology modeling. Due to many factors such as noise, conformational variability, experimental artifacts, and inexact model structures, existing automatic docking procedures are known to report false positives for a significant number of cases. The present paper focuses on a new technique to combine an offline exhaustive search algorithm with interactive visualization, collision detection, and haptic rendering. The resulting software system is highly immersive and allows the user to efficiently solve even difficult multi-resolution docking problems. Stereoscopic viewing, combined with head tracking and force feedback, generates an ideal virtual environment for true interaction with and solution of hybrid biomolecular modeling problems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
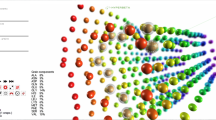
A key to understand the function of biological systems is the visualization of their natural state, ideally in a natural environment. At a molecular level, this is challenging. Traditional experimental techniques, such as X-ray crystallography, can provide the atomic structure of proteins, but only by removing them from their native surroundings and forcing them into a crystal lattice. Over the past decade, microscopy techniques have emerged as alternatives to these traditional structure determination methods, with the advantage of visualizing large multi-component molecules in a near-native state. Given the current focus of structural biology on interactions between proteins and better understanding of large protein complexes, cryo-electron microscopy (cryo-EM) has become a valuable tool (Frank 2002). Both image acquisition techniques and the computational synthesis of 3D volumetric models from micrographs have advanced considerably. 3D reconstructions of large protein complexes or even individual proteins can now be obtained (Fig. 1). While cryo-EM, thus, offers numerous advantages (small sample size, no need to crystallize, no packing effects, etc.), its main drawback is its inability to attain atomic resolution. Related techniques, such as cryo-electron tomography (cryo-ET; Lučić et al. 2005) and small-angle X-ray scattering (SAXS; Petoukhov and Svergun 2007), also yield non-atomic resolution 3D reconstructions of protein complexes and the methods discussed in this paper are equally applicable.

The interface of the Sculptor visualization package, showing a 3D volumetric reconstruction (from cryo-EM) of a GroEL chaperonin
The failure to achieve atomic resolution with cryo-EM, cryo-ET, or SAXS is not as big a stumbling block as one might expect. Often, crystal structures or good homology models are available for individual subunits. These known atomic structures can be docked into experimental volumetric reconstructions, yielding an atomic model of the whole complex (Steven and Baumeister 2008; Wriggers and Chacón 2001; Rossmann et al. 2005). Currently, various docking approaches exist, the results of which have lead to a significantly better understanding of the structure and interaction of proteins and protein complexes. However, the multi-resolution docking problem poses a series of challenges, which are detailed in Sect. 2. Those challenges cannot be dealt with by fully automated, exhaustive search-based, pattern matching algorithms. User input is vital and an immersive environment is key to allow the user to efficiently dock high-resolution structures into experimental volumetric data sets. The goal of our present work is to provide a fully immersive docking environment, which utilizes stereoscopic viewing, head tracking, and haptic rendering. Only such a truly interactive approach allows an in-depth, exhaustive, and efficient exploration of a given docking problem.
The present report first discusses both docking challenges and existing fitting methods, and then describes our novel technique.
2 Docking challenges and existing methods
The multi-resolution docking problem can be defined in the present context as the following: given a 3D volumetric map and a set of atomic structures, find the locations and orientations of the structures that best reproduce the volume data. Theoretically, maximizing a scoring function that measures the quality of the current docking will yield the desired result. Each structure should be fitted independently to avoid problems if not all atomic structures are known.
While the ultimate goal, stated above, seems feasible, numerous problems appear due to the nature of the experimental data sets. The actual micrographs obtained by cryo-EM contain large amounts of noise (Fig. 2), caused by the extremely low electron dose necessary to avoid destroying the specimen. The low signal-to-noise ratio can be improved by first aligning and then averaging particle projections of identical orientations. In practice, tens of thousands of individual particles images are required to produce a 3D reconstruction of 10 Å resolution or better (Frank 2006).

Six cryo-electron micrographs of the GroEL chaperonin
Several other factors also reduce the resolution or introduce distortions. First, no two protein complexes are truly identical due to small random conformational changes. Second, multiple stable conformations may exist, which differ significantly but are difficult to distinguish in the micrographs. Third, the density of the volume data is often heterogeneous, again caused by conformational fluctuations. Fourth, the orientational preferences of a biomolecule on the experimental support can lead to major gaps in the angular space of the projections and cause deformations in the 3D reconstruction. Finally, due to the complex nature of cryo-EM, artifacts may be introduced at any stage from the sample preparation to recording the actual micrograph.
The experimental difficulties of obtaining a good 3D reconstruction of a protein complex represent only one class of problems. Further challenges arise from the atomic structures used for the docking. X-ray structures may exhibit distortions due to crystal packing effects. Homology models are usually derived from different species and are only approximate models of the true atomic structure of the proteins under study.
All the above factors contribute in rendering multi-resolution docking an ill-posed problem. While the underlying theoretical problem is straightforward, including these complicating factors into a scoring function is difficult and may never be achieved. Therefore, the knowledge and reasoning of a human user will always be necessary to critically evaluate solutions to a given docking problem.
Two general approaches for solving the multi-resolution docking problem are currently prevalent: exhaustive evaluation of cross-correlation-based scoring functions (Chacón and Wriggers 2002; Jiang et al. 2001; Garzón et al. 2007; Volkmann and Hanein 1999; Roseman 2000; Rossmann 2000; Ceulemans and Russell 2004; Wu et al. 2003) or measuring the deviation of feature points of the multi-resolution 3D objects (Wriggers et al. 1999; Birmanns and Wriggers 2007). Each approach has different strengths and weaknesses—the method proposed in this work utilizes a cross-correlation coefficient as its main scoring function, an adaptation to feature-based scoring functions would also be possible.
In correlation-based docking, the scoring function is determined by projecting and blurring the atomic structure onto a 3D map and calculating the cross-correlation coefficient between this map and the original volume data. The statistical nature of cross-correlation ensures that high scores are generated not only for nearly perfect matches, but also for approximate agreement. This property is vital in the current context since near-perfect matches are rare. Using cross-correlation as a scoring function, the best docking locations are usually found via an exhaustive search. Such a docking procedure can be applied successfully to high-quality, intermediate resolution maps, but is prone to fail for more problematic data sets. In the past, efforts went into the development of advanced scoring functions that aim to counteract the artifacts of the experimental maps. An example is the introduction of an edge-detection filter like the Laplacian (Chacón and Wriggers 2002), which extends significantly the viable resolution range of automatic molecular-docking tools. Nevertheless, as stated above, the resolution is just one property of the volumetric map—numerous factors contribute to the final shape of the volume data and it is not feasible to model them accurately using a scoring function.
Besides the scoring function itself, exhaustive search methods encounter a second problem: How are candidate solutions picked? Solely relying on high docking scores is only practical in near-perfect data sets. In less optimal cases, many almost equivalent solutions exist and the user is inundated by hundreds of solutions for a protein complex containing only a few subunits.
To illustrate this point further, we chose the GroEL chaperonin (PDB entry 1GRL) as a test system. This protein is a homo-14mer with a monomer weight of approximately 47 kDa. We first performed a series of exhaustive searches at several resolutions. The atomic structure of 1GRL was low pass filtered, using a Gaussian kernel, to 10, 12.5, and 15 Å resolution. To simulate experimental density variations, the weight of atoms in the outer “rings” was varied between 1.00 and 0.70. This yielded a density decrease in these parts similar to that found in experimental maps (where it is due to motion of atoms in these regions). Using the simulated maps, 23 exhaustive searches were performed. For an automated search to succeed, the top 14 solution candidates have to correspond to the 14 monomers in the protein complex. As Table 1 shows, the exhaustive search can only be trusted at 10 Å resolution, with little or no density variation. These are the sole cases where the top 14 candidates represent the 14 monomers in the structure. At lower resolutions or larger density variations, the exhaustive search fails to produce a proper ranking of solutions. While all true solutions are still found, they are preceded by large numbers of false positives (see the “highest correct” column in Table 1).
As an alternative to the fully automatic docking tools, scientists often carry out purely user-guided docking in a visualization program (Kleywegt et al. 2001). This solely visual approach allows a biologist to directly apply his or her knowledge of the system under study. On the other hand, the procedure is highly subjective and the software does not support the user in any way. Manipulating a protein in six dimensions (6D, three translations and three rotations) is non-trivial and the best docking solutions are not necessarily evident.
In related scientific fields, virtual reality has been used successfully to enhance scientists’ understanding of a given problem and guide software towards promising solutions. Tactile feedback for drug-receptor docking simulations has been pioneered by Ouh-young et al. (1988). More recently, Bayazit et al. (2001) also used force feedback to guide a motion planning algorithm for ligand binding. In the present context of multi-resolution docking, our group has developed a haptic docking solution which computes a simplified scoring function and the related forces on the fly (Birmanns and Wriggers 2003).
Our current aim is to support the user with significantly more docking information and provide a fully immersive environment for solving challenging multi-resolution docking problems. Tactile clues about a scoring landscape are critical in such an endeavor, but they need to be based on an accurate scoring function. High-quality scoring functions are computationally expensive and therefore not suitable for real-time updates. We circumvent this problem by utilizing a fast exhaustive search pattern matching platform which can pre-compute a 3D field of fitting scores. Such a field is amicable to visualization, haptic rendering, and further feature extraction. Since the scoring field is calculated offline, complex, correlation based, scoring functions can be used and the interactive portion of our system can focus on generating additional docking information on the fly. One crucial piece of information is the interactions with already docked proteins. We developed an intuitive system which provides visual cues to the user about favorable interactions and highlights potential clashes. This combination of offline exhaustive search and online interaction detection allows an intuitive, visual, and haptic exploration of a given docking problem.
3 Interactive global docking
Interactive global docking (IGD) consists of two separate steps: first, an offline exhaustive search is performed in Eliquos, our new cross-correlation based exhaustive search software. This typically takes from a few minutes to a few hours, depending on the size of the system. The exhaustive search produces a vector field containing fitting scores and orientations of the probe structure, i.e., a scoring field. This field is read into our Sculptor visualization package and the user interactively explores both the scoring field and any additional docking information generated on-the-fly by Sculptor. Figure 3 shows an outline of the software architecture and the sections below discuss both steps in detail.

The overall software architecture of the interactive global docking system
3.1 Exhaustive search
Eliquos, which performs the offline exhaustive search, is a general multi-resolution docking application. It is geared towards fast and accurate docking of large data sets, using a scoring function based on cross-correlation. Several advanced filtering techniques are available to enable high-accuracy docking at both high and low resolutions. Like most exhaustive search docking programs, Eliquos is a black box in the sense that once the user specifies the input parameters, no further interaction occurs and the software will output a set of solutions guided solely by the chosen scoring function.
Eliquos is written in C, employing extensive application-specific low-level tuning, OpenMP, and the Intel Math Kernel Library (Intel Corporation 1994–2008) to achieve high performance. It uses the standard MRC file format (Short 2006) for volume data I/O and the PDB format (The worldwide Protein Data Bank 2008) for atomic structures. While the actual docking procedure follows the approach of Chacón and Wriggers (2002), Eliquos contains many improvements to increase the efficiency of the code. The two most prominent new features are pre-screening of possible orientations and a padding routine which extends the experimental map by the smallest amount necessary for Fast Fourier Transform (FFT) translational matching.
The six degrees of freedom (DOF) which are examined in a 6D exhaustive search are treated in two different ways. The translational DOF are explored via the application of the Fourier convolution theorem, which states that a convolution in real space is equivalent to a multiplication in Fourier space. Calculating the cross-correlation (CC) in Fourier space allows the simultaneous evaluation of the CC for all possible docking locations in a given map (using the voxels of the target map as an implicit grid). The three rotational DOF are scanned explicitly using a uniform angular grid. Typically, an angular grid resolution of under 10° is needed for sufficiently fine sampling, which leads to a large number of samples. At 9° angular step size, over 20,000 Euler angles need to be explored. Since the execution time of the docking algorithm scales linearly with the number of angles examined, it is judicious to prune the angular grid as much as possible. Eliquos achieves this pruning by testing each orientation and determining if the orientation is feasible from a purely geometric point of view. For example, the experimental map of a membrane protein will be much larger in one direction than in the other two, limiting the viable orientations of the equally elongated probe structure. On the other hand, when a small probe structure is docked into a very large protein complex, most (if not all) orientations need to be examined. Eliquos performs this screening procedure quickly by calculating the convex hull (Barber et al. 1996) of the probe structure. The convex hull describes an envelope around the probe and only contains the (small number of) atoms which span this envelope. The convex hull can, therefore, be used to rapidly determine which orientations will fit inside the experimental map.
Having found all feasible orientations, Eliquos then determines the amount of padding needed for the experimental volumetric map. Padding the map is necessary for several reasons: a given orientation might be viable but cannot fit into the map when rotated around the center of the probe structure, padding needed for filtering the map, padding to avoid FFT artifacts during the translational search, and padding to enable the use of the most efficient FFT pathways available. The actual amount of padding is determined taking all four factors into account. This approach ensures that only the absolute minimum of padding is added since the cost of the FFT scales as \({\mathcal{O}}(n \log n)\), where n is the total number of voxels.
In a final preparation step, a list of reasonable probe positions is generated. Again, purely geometric reasoning is applied to determine all possible positions of the probe structure. This step is necessary since Laplacian filtering, e.g., can generate false positive solutions, corresponding to exterior docking. In practice, Eliquos generates a spherical test volume with a diameter of half the smallest extent of the probe structure. A single unfiltered FFT cross-correlation calculation is then performed, which results in a 3D map of viable locations.
After all preparation steps are completed, the exhaustive 6D search can be performed (Fig. 4). The search loops over all feasible orientations, rotating and blurring the probe structure, and calculating the cross-correlation coefficients for all possible translations in the given orientation. At the end of each iteration, a list of candidate solutions is updated to reflect the currently best orientation at each position.

The main loop of the 6D exhaustive search in Eliquos
If a fully automated exhaustive search is desired, Eliquos then locates the highest ranked positions and corresponding orientations. The set of candidate solutions is sorted by docking score and then pruned to eliminate near-degenerate solutions. The resulting solutions are further optimized using a local optimization procedure. This off-grid optimization ensures the best possible docking of the high-resolution probe structure into the experimental 3D reconstruction.
In the case of IGD, Eliquos instead generates several files containing the scoring field. These files consist of two MRC volume data sets and one text file with a list of all sampled orientations. The first volume file contains the scalar component of the scoring field, i.e., the score of the best solution at each position. The second volume holds the corresponding orientation in the form of an index. The Sculptor visualization package can read in and use this scoring field as one of the docking criteria supporting the user’s fitting decisions.
3.2 Interactive visualization
The interactive stage of IGD is performed in the Sculptor visualization package. Sculptor (Fig. 1) is aimed primarily at working with volumetric data sets and docking of high-resolution structures into these 3D reconstructions. It makes extensive use of hardware graphics acceleration to provide high performance visualization of large data sets. Some of the provided features are as follows: volume manipulation tools, isosurface and direct volume rendering, feature-based multi-resolution docking using vector quantization, fast flexible fitting based on interpolation, and cross-correlation-based refinement of approximately fitted structures. In addition, Sculptor provides broad virtual reality support: stereoscopic rendering, head tracking, and various haptic devices are supported. The paragraphs below first describe the general software architecture of Sculptor and then outline the main loop of the IGD visualization phase. Finally, both the haptic rendering and steric interaction components are explained in more detail.
The application Sculptor is a thin software layer sitting on top of a general purpose scientific visualization and virtual reality library called SVT. SVT was developed for various in-house virtual reality projects, providing applications with a transparent software platform to access immersive, multi-display VR systems, and also conventional workstations. The C++ library combines a flexible VR rendering backend with advanced scientific visualization methods. Beside volume rendering and biomolecular visualization routines, the toolkit also includes support for tactile feedback using a variety of different haptic devices. The modules controlling the virtual reality and haptic rendering hardware are designed as a separate abstraction layer called LIVE. The LIVE layer provides a device-independent interface and permits flexible, on-demand loading of code. This way, devices can be added or removed after linking the main application, and even during run-time. In addition, the library makes VR devices accessible through the network and across different operating system platforms, without any changes in the application code.
Sculptor itself links SVT with biomolecular modeling routines and a Qt-based user interface. It thereby can run not only on most multi-display VR installations, but also on PC workstations typically found in experimental biology labs. If executed on a workstation, the user interface integrates the 3D rendering in the main application window (Fig. 1), to assure a similar user experience as in other 3D modeling packages. In a VR system, the 3D rendering area is detached from the user interface and shown on the display areas using a perspective correction based on the position information from the head-tracking device.
For IGD, Sculptor first loads the exhaustive search results generated by Eliquos. When IGD is activated, the main loop shown in Fig. 5 is entered. As the user moves the probe structure, Sculptor retrieves both the optimal orientation at the current position and the global docking score from the exhaustive search data. Then, the forces are calculated from the scoring field via quadratic interpolation. Next, steric interactions are determined and finally both the visual and haptic output is updated. Once a satisfactory probe position is found, it can be added to a set of candidate solutions. The user can easily switch the focus of IGD between the current position and any of the saved solutions, allowing an iterative refinement of the docking locations.

Sculptor: main loop during interactive global docking
Sculptor has already supported force-feedback guided docking for some time (Birmanns and Wriggers 2003). A local cross-correlation measure was used to give feedback about the local neighborhood and guide the user to favorable docking positions. This approach has the distinct advantage that no pre-computation is necessary and the user can start exploring the docking problem immediately. However, a computationally simple docking score must be used in this context, since haptic devices need real-time force updates in the order of 1,000 Hz to provide a smooth user experience. Full cross-correlation scores with advanced filtering are computationally too expensive. By using a pre-computed scoring field, scoring functions of arbitrary complexity can be used and the force calculation simplifies to a table lookup and trivial interpolation. The forces are determined via the first derivative of a quadratic polynomial, using the current score and its nearest neighbors as input data points for the interpolant. The interpolation and differentiation are performed separately along each principal axis. The orientations contained in the scoring field allow the software to rotate the probe structure into the best orientation at the current point in space. The user is thus only responsible for translating the structure, which greatly simplifies his task. Limiting the interactive search space to 3D also has the advantage that inexpensive haptic devices can be used for the force feedback. Novint Technologies, Inc. (2008), e.g., offers the Falcon 3D device priced below $200. While the Falcon was developed for immersive 3D gaming, it is ideally suited for our docking approach. The device only supports 3D translations but no rotations. In practice, the user experience with the Falcon is at least as good as, e.g., with a SensAble Phantom. Since only 3D translations are needed for IGD, the lack of rotational DOF is actually an advantage.
During the interactive stage of IGD, Sculptor not only uses the scoring field generated by Eliquos, but also generates additional, on-the-fly, docking information which the user can draw on to find ideal docking positions. The additional information is based on possible steric interactions between candidate solutions (i.e., favorable protein–protein distances). The aim is not to fully simulate complex protein–protein interactions but rather highlight areas of interest for the user. Whenever a new potential solution is found by the user, Sculptor calculates a 3D distance map which contains the distance from any point in space to the nearest backbone atom of all solutions. These distances are stored as a 3D volume where each voxel contains the corresponding distance. An example of such a map is shown in Fig. 6. The current candidate solutions are shown in light blue. The red and green volumes highlight forbidden and favorable positions for backbone atoms of the probe structure, respectively. This distance map allows a very efficient determination of good and bad protein interactions. The map is consulted at the positions of all C α atoms of the probe structure and the distance is compared to known interaction ranges.

Sculptor screenshot: example of a distance map used for efficient steric interaction determination. The current candidate solutions are shown in light blue. If any backbone atoms of the probe structure enter a red region, a steric clash with the existing solutions is likely. On the other hand, the green regions represent areas resulting in favorable interaction distances.
Källblad and Dean (2004) performed an extensive study of protein–protein contacts in nature. They concluded that only a narrow range of backbone–backbone distances occur at the interface between two proteins. Based on their results, we consider distances below 2.5 Å to represent a clash and 2.5–10 Å to be a favorable interaction. While these default values appear to be suitable for a wide range of docking problems, they can nonetheless be changed interactively.
4 Computational and VR details
Our VR setup is a self-built, low-cost back-projection system, employing polarization filters and inexpensive polarized glasses for stereoscopic viewing. Two DLP projectors with linear polarizing filters and a polarization preserving screen are used. Due to space constraints, two mirrors fold the light path, yielding a compact system with a 80 × 80 footprint and a 80 × 60 screen. An electro-magnetic head-tracking system is used to determine the viewer’s position and viewing direction. The projected view is continuously updated to provide the illusion of stationary 3D objects.
The display system is driven by an AMD Athlon64 4400+ X2 dual core processor with 2 GB of RAM and a NVidia Quadro FX 4500 graphics card. A SensAble Phantom 1.5/6DOF, connected via a parallel port, provides 6D input and force feedback. To garantee high haptic rendering update frequencies, the device control and force calculations are performed in a separate thread. Due to the pre-computed scoring field, this thread only consumes a fraction of one CPU core.
An inexpensive haptic device alternative for the desktop is the Novint Falcon. While not providing quite the fidelity of the Phantom, the Falcon is perfectly suitable for desktops or even laptops. Paired with visual force output via an arrow, it makes IGD also accessible on common and inexpensive hardware.
Sculptor, Eliquos, and the associated libraries are freely available in binary form from http://sculptor.biomachina.org. Executables for 32 and 64 bit Linux, Windows, Power and Intel Macs are available. Please contact the authors for access to the source code.
We also ported the exhaustive search export functionality into the established Colores tool (Chacón and Wriggers 2002) of the Situs docking package (available from http://situs.biomachina.org). This allows users to take advantage of IGD without requiring knowledge of the new Eliquos package.
5 IGD in practice
To give an example of docking with IGD, we return to the GroEL example given earlier. We start by loading the scoring field from the 15 Å resolution test with a density variation of 0.9 into Sculptor. The top scoring solution of the exhaustive search is shown in Fig. 7a and is clearly incorrect. As Table 1 shows, the first correct solution has a rank of 295, but the scoring field does contain all correct solutions. The IGD starting position (Fig. 7b) again reflects this high scoring but false solution. The probe structure can then be moved in 3D via a haptic device (Fig. 7c). The user is only responsible for translating the center of the monomer, the optimal orientation at the present position is determined by the scoring field. So, as the probe structure is moved around the experimental data set, it automatically rotates into the most favorable orientation. In addition, the (globally normalized) score at the present position is displayed graphically through color changes of the central sphere as well as numerically. The user then locates a suitable candidate position for the first docked monomer, taking the global docking score and his or her knowledge of the system into account. When trying to find such a position, which should be a local maximum in the scoring field, the force feedback becomes a crucial guide for the user and is further enhanced by visual feedback via a gradient arrow. When a local maximum is encountered, it is highlighted by a wire-frame overlay around the central sphere (Fig. 7d). Once a suitable location is chosen, it can be saved as a solution (Fig. 7e, white structure) and the probe is moved in search for the next candidate location. At this stage, the additional steric information generated by Sculptor comes into play. The user can visualize steric clashes between the current probe structure and all previously docked solutions (Fig. 7e) as well as good protein contacts (Fig. 7f). Once all constituent proteins are approximately placed, their positions can be further adjusted by either manual or automatic refinement. For example, Fig. 8 shows a closer view of the fit resulting from automatic Laplacian refinement of all subunits. No steric clashes are evident and the protein interfaces are highlighted by green spheres, signifying good contacts.

Stages of interactive global docking. The figure shows direct screen captures from the Sculptor visualization package. a Incorrect top scoring solution from exhaustive search. b Starting point of IGD. c Moving the probe. d Found a local maximum. e Steric clashes shown as red spheres. f Good contacts highlighted as green spheres

Results of automated refinement of the interactively docked structures. No steric clashes occur and the protein–protein interfaces are highlighted by green spheres
6 Interactive docking example: binding of Cdc6 to the origin recognition complex (ORC)
Binding of the Cdc6 protein to the ORC is a crucial step in the assembly of the DNA pre-replication complex (Liang et al. 1995) during cell division. The ORC–Cdc6 assembly has previously been extensively studied by Speck et al. (2005) using various techniques, including negative stain electron microscopy. The resulting volumetric data sets with 20 Å resolution are publicly available (EMDB entries 1156 and 1157). Figure 9a shows a map of the ORC by itself and Fig. 9b depicts the assembled ORC–Cdc6 complex. While a significant reorganization occurs in the ORC during assembly, Cdc6 clearly docks on the left side of the complex.

Interactive docking of ORC–Cdc6: a Experimental map of ORC without Cdc6. b Experimental map of ORC with Cdc6. c Globally best docking solution from exhaustive search. d Docking solution from IGD with subsequent local optimization.
Using PDB entry 1FNN (Liu et al. 2000) as the Cdc6 probe structure, we performed an exhaustive search with Laplacian filtering and refined the ten top scoring candidate solutions. An angular step size of 9° was used during the search, resulting in a search space of 20,400 angles. Figure 9c shows the ORC–Cdc6 complex with the top scoring docked solution (blue). However, this solution lies completely within the original ORC and can, therefore, not be valid. In fact, all the top ten solutions fall within the ORC part of the complex. These false positives are produced by the comparatively low resolution of 20 Å, a lack of interior details in negative stain maps, and an uneven density distribution in the experimental data.
Exploring the exhaustive search data via IGD in Sculptor yielded five candidate solutions in the correct area of the ORC–Cdc6 complex. A local optimization (again using Laplacian filtering) was performed on these solutions. The optimized candidates were ranked both via standard and Laplacian cross-correlation. Two of the candidates produce nearly identical top scores; however, one of them again showed significant overlap with the lone ORC structure. The remaining solution is shown in Fig. 9d and is similar to the manually docked structure by Speck et al.
The present example highlights the true power of IGD. Often, information about the rough position of a subunit is known from biology, but traditional exhaustive search methods fail to rank solutions in this area high enough. Thus, they are often not seen by the user. As mentioned above, uneven density distributions in the experimental data usually prevent automatic exhaustive search algorithms from succeeding in such cases. Picking the correct candidate solution then falls back on the user’s knowledge of the system’s biology and IGD greatly simplifies this process.
7 Conclusions
The IGD approach, presented here, combines the best features of non-interactive exhaustive search techniques and purely interactive visualization methods. It provides the user with both visual and haptic feedback about global docking scores, steric clashes, and good protein–protein interfaces. The additional information supplied during IGD allows biologists to not only rely on their personal knowledge of the system but also draw on objective, software-generated, fitting information. The currently available indicators represent only the first steps for incorporating more information into multi-resolution docking procedures. In the future, we plan to include contact information from mutation experiments, distances from NMR or FRET quenching measurements, as well as improving the existing scoring functions. Finally, the ORC–Cdc6 binding example demonstrates how our new approach allows for an efficient solution of a docking problem where traditional exhaustive search techniques fail. The highly immersive environment, provided by IGD, greatly facilitates this docking task for the user.
References
Barber CB, Dobkin DP, Huhdanpaa HT (1996) The quickhull algorithm for convex hulls. ACM Trans Math Software 22:469–483
Bayazit OB, Song G, Amato NM (2001) Ligand binding with OBPRM and haptic user input. In: 2001 IEEE international conference on robotics and automation. Seoul, Korea
Birmanns S, Wriggers W (2003) Interactive fitting augmented by force-feedback and virtual reality. J Struct Biol 144:123–131
Birmanns S, Wriggers W (2007) Multi-resolution anchor-point registration of biomolecular assemblies and their components. J Struct Biol 157:271–280
Ceulemans H, Russell RB (2004) Fast fitting of atomic structures to low-resolution electron density maps by surface overlap maximization. J Mol Biol 338:783–793
Chacón P, Wriggers W (2002) Multi-resolution contour-based fitting of macromolecular structures. J Mol Biol 317:375–384
Frank J (2002) Single-particle imaging of macromolecules by cryo-electron microscopy. Annu Rev Biophys Biomol Struct 31:303–319
Frank J (2006) Three-dimensional electron microscopy of macromolecular assemblies. Oxford University Press, New York
Garzón JI, Kovacs JA, Abagyan R, Chacón P (2007) ADP_EM: fast exhaustive multi-resolution docking for high-throughput coverage. Bioinformatics 23:427–433
Intel Corporation (1994–2008) Intel math kernel library 10.0
Jiang W, Baker ML, Ludtke SJ, Chiu W (2001) Bridging the information gap: computational tools for intermediate resolution structure interpretation. J Mol Biol 308:1033–1044
Källblad P, Dean PM (2004) Backbonebackbone geometry of tertiary contacts between α-helices. Proteins 56:693–703
Kleywegt GJ, Zou JY, Kjeldgaard M, Jones TA (2001) Around O. International tables for crystallography, vol F, chap 17.1. Kluwer, Dordrecht, pp 353–356, 366–367
Liang C, Weinreich M, Stillman B (1995) ORC and Cdc6p interact and determine the frequency of initiation of DNA replication in the genome. Cell 81:667–676
Liu J, Smith CL, DeRyckere D, DeAngelis K, Martin GS, Berger JM (2000) Structure and function of Cdc6/Cdc18: implications for origin recognition and checkpoint control. Mol Cell 6:637–648
Lučić V, Förster F, Baumeister W (2005) Structural studies by electron tomography: from cells to molecules. Annu Rev Biochem 74:833–865
Novint Technologies Inc. (2008) The Falcon haptic device. http://www.novint.com
Ouh-young M, Pique M, Hughes J, Srinivasan N, Brooks FP Jr (1988) Using a manipulator for force display in molecular docking. In: 1988 IEEE robotics and automation conference, vol 3. Philadelphia, PA, pp 1824–1829
Petoukhov MV, Svergun DI (2007) Analysis of X-ray and neutron scattering from biomacromolecular solutions. Curr Opin Struct Biol 17:562–571
Roseman AM (2000) Docking structures of domains into maps from cryo-electron microscopy using local correlation. Acta Crystallogr D 56:1332–1340
Rossmann MG (2000) Fitting atomic models into electron-microscopy maps. Acta Crystallogr D 56:1341–1349
Rossmann MG, Morais MC, Leiman PG, Zhang W (2005) Combining X-ray crystallography and electron microscopy. Structure 13:355–362
Short J (2006) MRC Cambridge Image Processing System 28 Sep 2006
Speck C, Chen Z, Li H, Stillman B (2005) ATPase-dependent cooperative binding of ORC and Cdc6 to origin DNA. Nat Struct Mol Biol 12:965–971
Steven AC, Baumeister W (2008) The future is hybrid. J Struct Biol 163:186–195
The worldwide Protein Data Bank (2008) Protein data bank contents guide: atomic coordinate entry format description
Volkmann N, Hanein D (1999) Quantitative fitting of atomic models into observed densities by electron microscopy. J Struct Biol 125:176–184
Wriggers W, Chacón P (2001) Modeling tricks and fitting techniques for multiresolution structures. Structure 9:779–788
Wriggers W, Milligan RA, McCammon JA (1999) Situs: a package for docking crystal structures into low-resolution maps from electron microscopy. J Struct Biol 125:185–195
Wu X, Milne JLS, Borgnia MJ, Rostapshov AV, Subramaniam S, Brooks BR (2003) A core-weighted fitting method for docking atomic structures into low-resolution maps: application to cryo-electron microscopy. J Struct Biol 141:63–76
Acknowledgments
We would like to thank Huilin Li for kindly discussing the ORC–Cdc6 results with us and Willy Wriggers for his valuable advice regarding this project. The present work was supported by a training fellowship (to J.H.) from the National Library of Medicine to the Keck Center for Interdisciplinary Bioscience Training of the Gulf Coast Consortia (NLM Grant No. 5T15LM- 07093), NIH grant R01GM62968, a grant from the Gillson-Longenbaugh Foundation, and startup funds from the University of Texas at Houston (to S.B.). In addition, we would like to thank the reviewers for many helpful comments and suggestions.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Heyd, J., Birmanns, S. Immersive structural biology: a new approach to hybrid modeling of macromolecular assemblies. Virtual Reality 13, 245–255 (2009). https://doi.org/10.1007/s10055-009-0129-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10055-009-0129-y