
Abstract:
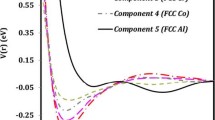
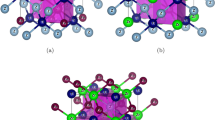
Results are presented of first-principles total-energy calculations and molecular-dynamics simulations of structural transformations in magnetic transition metal alloys like Fe1-xNix. While first-principles calculations allow to identify those structures having the lower total energy, molecular-dynamics simulations can be used to trace out the dependence of the transformation on temperature, composition, concentration of defects etc. We have used the method of the semi-empiric embedded-atom potential in the molecular-dynamics simulations which yields remarkable good results for the structural changes.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Received: 3 February 1998 / Revised: 4 May 1998 / Accepted: 24 June 1998
Rights and permissions
About this article
Cite this article
Entel, P., Meyer, R., Kadau, K. et al. Martensitic transformations: first-principles calculations combined with molecular-dynamics simulations. Eur. Phys. J. B 5, 379–388 (1998). https://doi.org/10.1007/s100510050457
Issue Date:
DOI: https://doi.org/10.1007/s100510050457