
Abstract
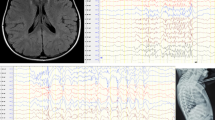
Developmental and epileptic encephalopathy (DEEs) (OMIM#618,328) is characterized by seizures, hypotonia, and brain abnormalities, often arising from mutations in genes crucial for brain function. Among these genes, GLS stands out due to its vital role in the central nervous system (CNS), with homozygous variants potentially causing DEE type 71. Using Whole Exome Sequencing (WES) on a patient exhibiting symptoms of epileptic encephalopathy, we identified a novel homozygous variant, NM_014905.5:c.1849G > T; p.(Asp617Tyr), in the GLS gene. The 5-year-old patient, born to consanguineous parents, presented with developmental delay, encephalopathy, frequent seizures, and hypotonia. Sanger sequencing further validated the GLS gene variant in both the patient and his family. Furthermore, our bioinformatics analysis indicated that this missense variant could lead to alteration of splicing, resulting in the activation of a cryptic donor site and potentially causing loss of protein function. Our finding highlights the pathogenic significance of the GLS gene, particularly in the context of brain disorders, specifically DEE71.


Similar content being viewed by others

Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- DEE71 :
-
Developmental and epileptic encephalopathy type 71;
- OMIM :
-
Online Mendelian inheritance in man;
- ACMG :
-
American College of Medical Genetics;
- WES :
-
Whole-exome sequencing;
- MAF :
-
Minor allele frequency;
- MRI :
-
Brain magnetic resonance imaging
References
Happ HC, Carvill GL (2020) A 2020 view on the genetics of developmental and epileptic encephalopathies. Epilepsy currents 20(2):90–96. https://doi.org/10.1177/153575972090611
Ramadan W, Patel N, Anazi S, Kentab A, Bashiri F, Hamad M et al (2017) Confirming the recessive inheritance of SCN1B mutations in developmental epileptic encephalopathy. Clin Genet 92(3):327–331. https://doi.org/10.1111/cge.12999
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS et al (2003) Human gene mutation database (HGMD®): 2003 update. Hum Mutat 21(6):577–581. https://doi.org/10.1002/humu.10212
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S et al (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44(D1):D862–D868. https://doi.org/10.1093/nar/gkv1222
Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31(13):3812–3814. https://doi.org/10.1093/nar/gkg509
Choi Y, Chan AP (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31(16):2745–2747. https://doi.org/10.1093/bioinformatics/btv195
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249. https://doi.org/10.1038/nmeth0410-248
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11(4):361–362. https://doi.org/10.1038/nmeth.2890
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T et al (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44(W1):W344–W350. https://doi.org/10.1093/nar/gkw408
Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu Y et al (2003) The UCSC genome browser database. Nucleic Acids Res 31(1):51–54. https://doi.org/10.1093/nar/gkg129
Esmaeilzadeh-Gharehdaghi E, Razmara E, Bitaraf A, Mahmoudi M, Garshasbi M. (2019) S3440P substitution in C-terminal region of human Reelin dramatically impairs secretion of Reelin from HEK 293T cells. Cellular and Molecular Biology. 65(6):12–6. https://doi.org/10.14715/cmb/2019.65.6.3
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F (2010) Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26(16):2069–2070. https://doi.org/10.1093/bioinformatics/btq330
Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S (2008) The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Human Genet 83(5):610–615. https://doi.org/10.1016/j.ajhg.2008.09.017
Thorvaldsdóttir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14(2):178–192. https://doi.org/10.1093/bib/bbs017
Márquez J, de la Oliva ARL, Matés JM, Segura JA, Alonso FJ (2006) Glutaminase: a multifaceted protein not only involved in generating glutamate. Neurochem Int 48(6–7):465–471. https://doi.org/10.1016/j.neuint.2005.10.015
Pérez-Gómez C, Matés JM, Gómez-Fabre PM, CASTILLO-OLIVARES Ad, Alonso FJ, Márquez J. (2003) Genomic organization and transcriptional analysis of the human L-glutaminase gene. Biochemical Journal. 370(3):771–84. https://doi.org/10.1042/bj20021445
Rumping L, Büttner B, Maier O, Rehmann H, Lequin M, Schlump J-U et al (2019) Identification of a loss-of-function mutation in the context of glutaminase deficiency and neonatal epileptic encephalopathy. JAMA Neurol 76(3):342–350. https://doi.org/10.1001/jamaneurol.2018.2941
Feng Y, Zhang C, Wei Z, Li G, Gan Y, Liu C et al (2022) Gene variations of glutamate metabolism pathway and epilepsy. Acta Epileptologica 4(1):1–15. https://doi.org/10.1186/s42494-022-00103-2
Rumping L, Tessadori F, Pouwels PJ, Vringer E, Wijnen JP, Bhogal AA et al (2019) GLS hyperactivity causes glutamate excess, infantile cataract and profound developmental delay. Hum Mol Genet 28(1):96–104. https://doi.org/10.1093/hmg/ddy330
Rumping L, Pouwels PJ, Wolf NI, Rehmann H, Wamelink MM, Waisfisz Q et al (2023) A second case of glutaminase hyperactivity: Expanding the phenotype with epilepsy. JIMD reports 64(3):217–222. https://doi.org/10.1002/jmd2.12359
Acknowledgements
We are very thankful to the patient’s family members for collaborating in this study, and also thankful to the staff of the DeNA laboratory, in Tehran, Iran, for helping us in this study.
Funding
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
A.B. and M.G. designed the basic concept of the research. A.R.T. carried out sample collection and clinical examinations. A.B., M.A.GH., and S.M.K. coordinated the genetic tests, the raw data and bioinformatics analysis. A.B. and M.A.GH. wrote the manuscript with valuable scientific commentary and input from all authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Ethics Committee of Children's Medical Center Hospital, Tehran, Iran approved the study and was conducted according to the ethical principle of the Declaration of Helsinki.
Consent to participate
We obtained written informed consent from all family participants. Specifically, the parents of the minor patient (5 years old) provided signed informed consent forms for their participation in clinical and genetic research.
Competing interests
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bazgir, A., Agha Gholizadeh, M., Kahani, S.M. et al. Identification of a Novel Homozygous GLS Gene Variant Associated with Developmental and Epileptic Encephalopathy (DEE) Type 71. Neurogenetics (2024). https://doi.org/10.1007/s10048-024-00753-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10048-024-00753-z