
Abstract
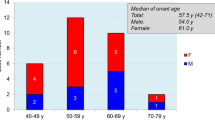
In genetic prion diseases (gPrD), five genetic variants (E200K, V210I, V180I, P102L, and D178N) are responsible for about 85% of cases. The R208H is one of the several additional rare mutations and to date, only 16 cases carrying this mutation have been reported worldwide. To describe the phenotypic features of 5 affected patients belonging to apparently unrelated Sardinian (Italian) families with R208H gPrD, and provide evidence for a possible founder effect are the aims of this study. The R208H PRNP mutation has a much higher relative frequency in Sardinia than elsewhere in Italy (72% vs. 4.4% of gCJD cases). Our cohort shared similar phenotypic features to the previously described patients with R208H-129M haplotype with most patients showing the classical Creutzfeldt-Jakob disease (CJD) phenotype. The analysis of 10 controls and 5 patients by NGS sequencing identified 4 haplotypes, 3 associated with the wild type variant, and one (H1) shared by all patients carrying the 208His variant. This is the first report of a regional cluster for R208H mutation in gPrD and the first report of the presence of a common ancestor for this Sardinian R208H cluster, confirming the probable consequences of genetic isolation process even for rare diseases.

Similar content being viewed by others

Data availability
The authors confirm that the data supporting the findings of this study are available within the article. If needed, further data that support the findings of this study are available from the corresponding author, [MM], upon reasonable request.
References
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144
Takada LT, Kim MO, Cleveland RW, Wong K, Forner SA, Gala II, Fong JC, Geschwind MD (2017) Genetic prion disease: experience of a rapidly progressive dementia center in the United States and a review of the literature. Am J Med Genet B Neuropsychiatr Genet 174:36–69
Ladogana A. and Gabor G. Kovacs (2018) Handbook of clinical neurology, Vol. 153 (3rd series) Human Prion Diseases. M. Pocchiari and J. Manson, Editors
Ladogana A, Puopolo M, Poleggi A, Almonti S, Mellina V, Equestre M, Pocchiari M (2005) High incidence of genetic human transmissible spongiform encephalopathies in Italy. Neurology 64:1592–1597
Minikel EV, Sonia M, Vallabh SM, Lek M et al (2016) Quantifying penetrance in a dominant disease gene using large population control cohorts. Sci Transl Med 8(322):322ra9. https://doi.org/10.1126/scitranslmed Aad 5169
Rodríguez-Martínez AB, Alfonso-Sánchez MA, Peña JA, Sánchez-Valle R, Zerr I, Capellari S, Calero M, Zarranz JJ, de Pancorbo MM (2008 May) Molecular evidence of founder effects of fatal familial insomnia through SNP haplotypes around the D178N mutation. Neurogenetics. 9(2):109–118. https://doi.org/10.1007/s10048-008-0120-x
McGuire LI, Poleggi A, Poggiolini I et al (2016) Cerebrospinal fluid real-time quaking-induced conversion is a robust and reliable test for sporadic creutzfeldt-jakob disease: an international study. Ann Neurol 80(1):160–165. https://doi.org/10.1002/ana.24679
Mackenzie G, Will R (2017) Creutzfeldt-Jakob disease: recent developments. F1000Res 6:2053
Poleggi A, van der Lee S, Capellari S et al (2018) Age at onset of genetic (E200K) and sporadic Creutzfeldt-Jakob diseases is modulated by the CYP4X1 gene. J Neurol Neurosurg Psychiatry 89:1243–1249
Lattanzio F, Abu-Rumeileh S, Franceschini A et al (2017) Prion-specific and surrogate CSF biomarkers in creutzfeldt-Jakob disease: diagnostic accuracy in relation to molecular subtypes and analysis of neuropathological correlates of p-tau and Abeta42 levels. Acta Neuropathol 133(4):559–578
National UK CJD Research & surveillance unit protocol: https://www.cjd.ed.ac.uk/sites/default/files/criteria.pdf
Mastrianni JA, Iannicola C, Myers RM et al (1996) Mutation in prion protein gene at codon 208 in familial Creutzfeldt-Jakob disease. Neurology 47:1305
Roeber S, Krebs B, Neumann EM et al (2005) Creutzfeldt-Jakob disease in a patient with an R208H mutation of the prion protein gene (PRNP) and a 17-kDa prion protein fragment. Acta Neuropathol 109:443–448. https://doi.org/10.1007/s00401-004-0978-0
Nozaki I, Hamaguchi T, Sanjo N et al (2010) Prospective 10-year surveillance of human prion diseases in Japan. Brain 133:3043–3057
Chen C, Shi Q, Tian C et al (2011) The first Chinese case of Creutzfeldt-Jakob disease patient with R208H mutation in PRNP. Prion 5(3):232–234
Shi Q, Zhou W, Chen C et al (2015) The features of genetic prion diseases based on Chinese surveillance program. PLoS One 10(10):e0139552
Basset-Leobon C, Uro-Coste E, Peoc’h K, Haik S, Sazdovitch V, Rigal M, Andreoletti O, Hauw JJ, Delisle MB (2006) Familial Creutzfeldt-Jakob disease with an R208H-129V haplotype and Kuru plaques. Arch Neurol 63:449–452
Matej R, Kovacs GG, Johanidesova S et al (2012) Genetic Creutzfeldt–Jakob disease with R208H mutation presenting as progressive supranuclear palsy. Mov Disord 27(4):476–479. https://doi.org/10.1002/mds.24002
Mitrova E, Belay G, Slivarichova-Zakova D, Stelzer M (2015) The first case of genetic CJD with the rare mutation R208H, Met/Val heterozygous at codon 129 of the prion protein gene. Clin Med J 1(3):101–105
Capellari S, Cardone F, Notar S et al (2005) Creutzfeldt–Jakob disease associated with the R208H mutation in the prion protein gene. Neurology 64:905–907
Tiple D, Poleggi A, Mellina V et al (2019) Clinicopathological features of the rare form of Creutzfeldt-Jakob disease in R208H-V129V PRNP carrier. Acta Neuropathol Commun 7:47. https://doi.org/10.1186/s40478-019-0699-1
Vita MG, Gaudino S, Di Giuda D et al (2013) R208H-129VV haplotype in the prion protein gene: phenotype and neuroimaging of a patient with genetic Creutzfeldt-Jakob disease. J Neurol 260:2650–2652. https://doi.org/10.1007/s00415-013-7078-9
Appleby BS, Appleby KK, Crain BJ, Onyike CU, Wallin MT, Rabins PV (2009) Characteristics of established and proposed sporadic Creutzfeldt-Jakob disease variants. Arch Neurol 66(2):208–215. https://doi.org/10.1001/archneurol.2008.533
Piazza A, Mayr WR, Contu L et al (1985) Genetic and population structure of four Sardinian villages. Ann Hum Genet 49(pt 1):47–63
Contu D, Morelli L, Santoni F et al (2008) Y-chromosome based evidence for pre-neolithic origin of the genetically homogeneous but diverse Sardinian population: inference for association scans. PLoS One 3(1):e1430
Lee HS, Sambuughin N, Cervenakova L, Chapman J, Pocchiari M, Litvak S, Qi HY, Budka H, del Ser T, Furukawa H, Brown P, Gajdusek DC, Long JC, Korczyn AD, Goldfarb LG (1999 Apr) Ancestral origins and worldwide distribution of the PRNP 200K mutation causing familial Creutzfeldt-Jakob disease. Am J Hum Genet 64(4):1063–1070
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Melis, M., Molari, A., Floris, G. et al. Genetic Creutzfeldt-Jakob disease in Sardinia: a case series linked to the PRNP R208H mutation due to a single founder effect. Neurogenetics 21, 251–257 (2020). https://doi.org/10.1007/s10048-020-00618-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-020-00618-1