
Abstract
Context
In recent decades, drug development has become extremely important as different new diseases have emerged. However, drug discovery is a long and complex process with a very low success rate, and methods are needed to improve the efficiency of the process and reduce the possibility of failure. Among them, drug design from scratch has become a promising approach. Molecules are generated from scratch, reducing the reliance on trial and error and prefabricated molecular repositories, but the optimization of its molecular properties is still a challenging multi-objective optimization problem.
Methods
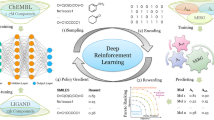
In this study, two stack-augmented recurrent neural networks were used to compose a generative model for generating drug-like molecules, and then reinforcement learning was used for optimization to generate molecules with desirable properties, such as binding affinity and the logarithm of the partition coefficient between octanol and water. In addition, a memory storage network was added to increase the internal diversity of the generated molecules. For multi-objective optimization, we proposed a new approach which utilized the magnitude of different attribute reward values to assign different weights to molecular optimization. The proposed model not only solves the problem that the properties of the generated molecules are extremely biased towards a certain attribute due to the possible conflict between the attributes, but also improves various properties of the generated molecules compared with the traditional weighted sum and alternating weighted sum, among which the molecular validity reaches 97.3%, the internal diversity is 0.8613, and the desirable molecules increases from 55.9 to 92%.






Similar content being viewed by others


Data availability
All data are available for download, for example, the ChEMBL226 dataset is available for download at https://www.ebi.ac.uk/chembl/g/#browse/activities/filter/target_chembl_id%3ACHEMBL226, the ChEMBL237 dataset is available for download at https://www.ebi.ac.uk/chembl/g/#browse/activities/filter/target_chembl_id%3ACHEMBL237.
Code Availability
All python code for this study is freely available at https://github.com/PengWeiHu1/mul_RL/tree/master
References
Schmidhuber J (2015) Deep learning in neural networks: an overview. Neural Netw 61:85–117. https://doi.org/10.1016/j.neunet.2014.09.003
Weininger D (1988) SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Model 28:31–36. https://doi.org/10.1021/ci00057a005
Santos BP, Abbasi M, Pereira T et al (2021) Optimizing recurrent neural network architectures for de novo drug design. 34th International Symposium on Computer-Based Medical Systems (CBMS). IEEE, pp 172–177. https://doi.org/10.1109/CBMS52027.2021.00067
Zheng S, Yan X, Gu Q et al (2019) QBMG: quasi-biogenic molecule generator with deep recurrent neural network. J Cheminf 11:1–12. https://doi.org/10.1186/s13321-019-0328-9
Arús-Pous J, Johansson SV, Prykhodko O et al (2019) Randomized SMILES strings improve the quality of molecular generative models. J Cheminf 11:1–13. https://doi.org/10.1186/s13321-019-0393-0
Hochreiter S, Schmidhuber J (1997) Long short-term memory. Neural Comput 9:1735–1780. https://doi.org/10.1007/978-3-642-24797-2-4
Blum LC, Reymond JL (2009) 970 million druglike small molecules for virtual screening in the chemical universe database GDB-13. J Am Chem Soc 131:8732–8733. https://doi.org/10.1021/ja902302h
Blaschke T, Olivecrona M, Engkvist O et al (2018) Application of generative autoencoder in de novo molecular design. Mol Inform 37:1–2. https://doi.org/10.1002/minf.201700123
Sutton RS, Barto AG (2018) Reinforcement learning: an introduction. MIT press
Zhou Z, Kearnes S, Li L et al (2019) Optimization of molecules via deep reinforcement learning. Sci Rep 9:1–10. https://doi.org/10.1038/s41598-019-47148-x
Jeon W, Kim D (2020) Autonomous molecule generation using reinforcement learning and docking to develop potential novel inhibitors. Sci Rep-uk 10:1–11. https://doi.org/10.1038/s41598-020-78537-2
Silver D, Huang A, Maddison CJ et al (2016) Mastering the game of Go with deep neural networks and tree search. Nature 529:484–489. https://doi.org/10.1038/nature16961
Pereira T, Abbasi M, Ribeiro B et al (2021) Diversity oriented deep reinforcement learning for targeted molecule generation. J Cheminf 13:1–17. https://doi.org/10.1186/s13321-021-00498-z
Olivecrona M, Blaschke T, Engkvist O et al (2017) Molecular de-novo design through deep reinforcement learning. J Cheminf 9:1–14. https://doi.org/10.1186/s13321-017-0235-x
Liu XH, Ye K, van Vlijmen HW et al (2019) An exploration strategy improves the diversity of de novo ligands using deep reinforcement learning: a case for the adenosine A2A receptor. J Cheminf 11:1–16. https://doi.org/10.1186/s13321-019-0355-6
Williams RJ (1992) Simple statistical gradient-following algorithms for connectionist reinforcement learning. Mach Learn 8:229–256. https://doi.org/10.1007/978-1-4615-3618-5-2
Panteleev J, Gao H, Jia L et al (2018) Recent applications of machine learning in medicinal chemistry. Bioorg Med Chem Lett 28:2807–2815. https://doi.org/10.1016/j.bmcl.2018.06.046
Mayer G, Heckel A (2006) Biologically active molecules with a “light switch”. Angew Chem Int Edit 45:4900–4921. https://doi.org/10.1002/anie.200600387
Khemchandani Y, O’Hagan S, Samanta S et al (2020) DeepGraphMolGen, a multi-objective, computational strategy for generating molecules with desirable properties: a graph convolution and reinforcement learning approach. J Cheminf 12:1–17. https://doi.org/10.1186/s13321-020-00454-3
Preuer K, Renz P, Unterthiner T et al (2018) Fréchet ChemNet distance: a metric for generative models for molecules in drug discovery. J Chem Inf Model 58:1736–1741. https://doi.org/10.1021/acs.jcim.8b00234
Benhenda M (2018) Can AI reproduce observed chemical diversity? BioRxiv. 292177. https://doi.org/10.1101/292177
Pereira T, Abbasi M, Oliveira JL et al (2021) Optimizing blood–brain barrier permeation through deep reinforcement learning for de novo drug design. Bioinformatics 37:i84–i92. https://doi.org/10.1093/bioinformatics/btab301
Sweeney MD, Sagare AP, Zlokovic BV et al (2018) Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14:133–150. https://doi.org/10.1038/nrneurol.2017.188
Liu F, Zhang L, Jin Z et al (2020) Modeling programs hierarchically with stack-augmented LSTM. J Syst Software 164:110547. https://doi.org/10.1016/j.jss.2020.110547
Tropsha A (2010) Best practices for QSAR model development, validation, and exploitation. Mol Inform 29:476–488. https://doi.org/10.1002/minf.201000061
Chen JF, Eltzschig HK, Fredholm BB et al (2013) Adenosine receptors as drug targets—what are the challenges? Nat Rev Drug Discov 12:265–286. https://doi.org/10.1038/nrd3955
Shang Y, Filizola M (2015) Opioid receptors: Structural and mechanistic insights into pharmacology and signaling. Eur J Pharmacol 763:206–213. https://doi.org/10.1016/j.ejphar.2015.05.012
Blaschke T, Engkvist O, Bajorath J et al (2020) Memory-assisted reinforcement learning for diverse molecular de novo design. J Cheminf. 12:1–17. https://doi.org/10.1186/s13321-020-00473-0
Goel M, Raghunathan S, Laghuvarapu S et al (2021) MoleGuLAR: molecule generation using reinforcement learning with alternating rewards. J Chem Inf Model 61:5815–5826. https://doi.org/10.1021/acs.jcim.1c01341
Grisoni F, Moret M, Lingwood R et al (2020) Bidirectional molecule generation with recurrent neural networks. J Chem Inf Model 60:1175–1183. https://doi.org/10.1021/acs.jcim.9b00943
Elton DC, Boukouvalas Z, Fuge MD et al (2019) Deep learning for molecular design—a review of the state of the art. Mol Syst Des Eng 4:828–849. https://doi.org/10.1039/C9ME00039A
Popova M, Isayev O, Tropsha A et al (2018) Deep reinforcement learning for de novo drug design. Sci Adv 4:eaap7885. https://doi.org/10.1126/sciadv.aap7885
Ertl P, Schuffenhauer A (2009) Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J Cheminformatic 1:1–11. https://doi.org/10.1186/1758-2946-1-8
Bickerton GR, Paolini GV, Besnard J et al (2012) Quantifying the chemical beauty of drugs. Nat Chem 4:90–98. https://doi.org/10.1038/nchem.1243
Leo A, Hansch C, Elkins D (1971) Partition coefficients and their uses. Chem Rev 71:525–616. https://doi.org/10.1021/cr60274a001
Polykovskiy D, Zhebrak A, Sanchez-Lengeling B et al (2020) Molecular sets (MOSES): a benchmarking platform for molecular generation models. Front Pharmacol 11:565644. https://doi.org/10.3389/fphar.2020.565644
Bento AP, Gaulton A, Hersey A et al (2014) The ChEMBL bioactivity database: an update. Nucleic Acids Res 42:D1083–D1090. https://doi.org/10.1093/nar/gkt1031
Gramatica P, Sangion A (2016) A historical excursus on the statistical validation parameters for QSAR models: a clarification concerning metrics and terminology. J Chem Inf Model 56:1127–1131. https://doi.org/10.1021/acs.jcim.6b00088
Benet LZ, Hosey CM, Ursu O et al (2016) BDDCS, the rule of 5 and drugability. Adv Drug Deliv Rev 101:89–98. https://doi.org/10.1016/j.addr.2016.05.007
Sanchez-Lengeling B, Outeiral C, Guimaraes GL et al (2017) Optimizing distributions over molecular space. An objective-reinforced generative adversarial network for inverse-design chemistry (ORGANIC). https://doi.org/10.26434/chemrxiv.5309668.v3
Funding
This work was supported by the National Natural Science Foundation of China (12261060 and 21665016), and the Natural Science Foundation of Jiangxi Province (20192BAB204010).
Author information
Authors and Affiliations
Contributions
P.H., J.Z., J.Y., and S.S. contributed to the study conception, design and analysis. Material preparation and data collection were performed by P.H. The first draft of the manuscript was written by P.H. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 595 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hu, P., Zou, J., Yu, J. et al. De novo drug design based on Stack-RNN with multi-objective reward-weighted sum and reinforcement learning. J Mol Model 29, 121 (2023). https://doi.org/10.1007/s00894-023-05523-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05523-6