
Abstract
Ribosomal protein S6 kinase beta-1 (S6K1) is considered a potential target for the treatment of various diseases, such as obesity, type II diabetes, and cancer. Development of novel S6K1 inhibitors is an urgent and important task for the medicinal chemists. In this research, an effective ensemble-based virtual screening method, including common feature pharmacophore model, 3D-QSAR pharmacophore model, naïve Bayes classifier model, and molecular docking, was applied to discover potential S6K1 inhibitors from BioDiversity database with 29,158 compounds. Finally, 7 hits displayed considerable properties and considered as potential inhibitors against S6K1. Further, carefully analyzing the interactions between these 7 hits and key residues in the S6K1 active site, and comparing them with the reference compound PF-4708671, it was found that 2 hits exhibited better binding patterns. In order to further investigate the mechanism of the interactions between 2 hits and S6K1 at simulated physiological conditions, the molecular dynamics simulation was performed. The ΔGbind energies for S6K1-Hit1 and S6K1-Hit2 were − 111.47 ± 1.29 and − 54.29 ± 1.19 kJ mol−1, respectively. Furthermore, deep analysis of these results revealed that Hit1 was the most stable complex, which can stably bind to S6K1 active site, interact with all of the key residues, and induce H1, H2, and M-loop regions changes. Therefore, the identified Hit1 may be a promising lead compound for developing new S6K1 inhibitor for various metabolic diseases treatment.
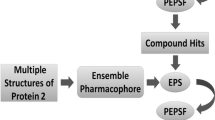
Graphical abstract














Similar content being viewed by others

Data availability
The data and materials are available from the corresponding authors on reasonable request.
References
Fenton TR et al (2011) Functions and regulation of the 70 kDa ribosomal S6 kinases. Int J Biochem Cell B 43:47–59. https://doi.org/10.1016/j.biocel.2010.09.018
Shamji AF et al (2003) Integration of growth factor and nutrient signaling: implications for cancer biology. Mol Cell 12:271–280. https://doi.org/10.1016/j.molcel.2003.08.016
Magnuson B et al (2012) Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J 441:1–21. https://doi.org/10.1042/BJ20110892
Couty S et al (2013) The discovery of potent ribosomal S6 kinase inhibitors by high-throughput screening and structure-guided drug design. Oncotarget 4:1647. https://doi.org/10.18632/oncotarget.1255
Zaiets I et al. (2018) The p60-S6K1 isoform of ribosomal protein S6 kinase 1 is a product of alternative mRNA translation, UBJ 25–35, https://doi.org/10.15407/ubj90.04.025
Yin Y et al (2020) Computer-aided discovery of phenylpyrazole based amides as potent S6K1 inhibitors. RSC Med Chem 11:583–590. https://doi.org/10.1039/C9MD00537D
Dann SG et al (2007) mTOR Complex1–S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med 13:252–259. https://doi.org/10.1016/j.molmed.2007.04.002
Patra T et al (2021) Inhibition of p70 isoforms of S6K1 induces anoikis to prevent transformed human hepatocyte growth. Life Sci 265:118764. https://doi.org/10.1016/j.lfs.2020.118764
Bussenius J et al (2012) Design and evaluation of a series of pyrazolopyrimidines as p70S6K inhibitors. Bioorg Med Chem Lett 22:2283–2286. https://doi.org/10.1016/j.bmcl.2012.01.105
Dhar R et al (2008) Constitutive activation of p70 S6 kinase is associated with intrinsic resistance to cisplatin. Int J Mol Sci 32:1133–1137. https://doi.org/10.3892/ijo.32.5.1133
Assad DX et al (2018) Additive cytotoxic effects of radiation and mTOR inhibitors in a cervical cancer cell line. Pathok Res Pract 214:259–262. https://doi.org/10.1016/j.prp.2017.10.019
Xie G et al (2017) Dual blocking of PI3K and mTOR signaling by NVP-BEZ235 inhibits proliferation in cervical carcinoma cells and enhances therapeutic response. Cancer Lett 388:12–20. https://doi.org/10.1016/j.canlet.2016.11.024
Nam KH et al (2019) Identification of a novel S6K1 inhibitor, rosmarinic acid methyl ester, for treating cisplatin-resistant cervical cancer. BMC Cancer 19:1–13. https://doi.org/10.1186/s12885-019-5997-2
Pearce LR, Alton GR et al (2010) Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem J 431:245–255. https://doi.org/10.1042/BJ20101024
Wang J, Zhong C et al (2013) Crystal structures of S6K1 provide insights into the regulation mechanism of S6K1 by the hydrophobic motif. Biochem J 454:39–47. https://doi.org/10.1042/BJ20121863
Tolche A, Goldman J et al (2014) A phase I trial of LY2584702 tosylate, a p70 S6 kinase inhibitor, in patients with advanced solid tumours. Eur J Cancer 50:67–75. https://doi.org/10.1016/j.ejca.2013.11.039
Hollebecque A, Houédé N et al (2014) A phase Ib trial of LY2584702 tosylate, a p70 S6 inhibitor, in combination with erlotinib or everolimus in patients with solid tumours. Eur J Cancer 50:76–84. https://doi.org/10.1016/j.ejca.2013.12.006
Morreale A, Mallon B et al (1997) Ro31–8220 inhibits protein kinase C to block the phorbol ester-stimulated release of choline- and ethanolamine-metabolites from C6 glioma cells: p70 S6 kinase and MAPKAP kinase-1beta do not function downstream of PKC in activating PLD. FEBS Lett 417:38–42. https://doi.org/10.1016/s0014-5793(97)01252-0
Couty S, Westwood IM et al (2013) The discovery of potent ribosomal S6 kinase inhibitors by high-throughput screening and structure-guided drug design. Oncotarget 4:1647–1661. https://doi.org/10.18632/oncotarget.1255
Bae EJ, Yang YM et al (2007) Identification of a novel class of dithiolethiones that prevent hepatic insulin resistance via the adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway. Hepatology 46:730–739. https://doi.org/10.1002/hep.21769
Ye P et al (2011) Potent and selective thiophene urea-templated inhibitors of S6K. Bioorg Med Chem Lett 21:849–852. https://doi.org/10.1016/j.bmcl.2010.11.069
Bussenius J et al (2012) Design and evaluation of a series of pyrazolopyrimidines as p70S6K inhibitors. Bioorg Med Chem Lett 22(6):2283–2286. https://doi.org/10.1016/j.bmcl.2012.01.105
Ip CKM et al (2012) Exploiting p70 S6 kinase as a target for ovarian cancer. Expert Opin Ther Targets 16(6):619–630. https://doi.org/10.1517/14728222.2012.684680
Chi OZ et al (2019) Inhibition of p70 ribosomal S6 kinase 1 (S6K1) by PF-4708671 decreased infarct size in early cerebral ischemia-reperfusion with decreased BBB permeability. Eur J Pharmacol 855:202–207. https://doi.org/10.1016/j.ejphar.2019.05.010
Estridge TB et al (2017) Identification of 4-aminopyrazolopyrimidine metabolite that may contribute to the hypolipidemic effects of LY2584702 in Long Evans diet–induced obese rats. J Pharmacol Exp Ther 362:108–118. https://doi.org/10.1124/jpet.117.240242
Zhang N et al (2020) Research progress of 70 kDa ribosomal protein S6 kinase (P70S6K) inhibitors as effective therapeutic tools for obesity, type II diabetes and cancer. Curr Med Chem 27:4699–4719. https://doi.org/10.2174/0929867327666200114113139
Baron R et al. (2012) Computational drug discovery and design, translated book computational drug discovery and design. https://doi.org/10.1007/978-1-61779-465-010.3390/molecules18010735
Anderson AC (2003) The process of structure-based drug design. Chem Biol 10:787–797. https://doi.org/10.1016/j.chembiol.2003.09.002
Shoichet BK (2004) Virtual screening of chemical libraries. Nature 432:862–865. https://doi.org/10.1038/nature03197
Singh N et al (2021) Virtual screening web servers: designing chemical probes and drug candidates in the cyberspace. Brief Bioinform 22:1790–1818. https://doi.org/10.1093/bib/bbaa034
Lin X et al (2020) A review on applications of computational methods in drug screening and design. Molecules 25:1375. https://doi.org/10.3390/molecules25061375
D’Souza S et al (2020) Machine learning models for drug–target interactions: current knowledge and future directions. Drug Discov Today 25:748–756. https://doi.org/10.1016/j.drudis.2020.03.003
Zhu J et al (2021) A multi-conformational virtual screening approach based on machine learning targeting PI3Kγ. Mol Divers 25:1271–1282. https://doi.org/10.1007/s11030-021-10243-1
Pinzi L et al (2019) Molecular docking: shifting paradigms in drug discovery. Int J Mol Sci 20:4331. https://doi.org/10.3390/ijms20184331
Niwa H et al (2014) Crystal structures of the S6K1 kinase domain in complexes with inhibitors. J Struct Funct Genomics 15:153–164. https://doi.org/10.1007/s10969-014-9188-8
Adewumi AT et al (2020) Thompson loop: opportunities for antitubercular drug design by targeting the weak spot in demethylmenaquinone methyltransferase protein. RSC Adv 10:23466–23483. https://doi.org/10.1039/D0RA03206A
Batool M, Ahmad B, Choi S (2019) A structure-based drug discovery paradigm. Int J Mol Sci 20:2783. https://doi.org/10.3390/ijms20112783
Zhang H et al (2022) Discovery of novel microtubule stabilizers targeting taxane binding site by applying molecular docking, molecular dynamics simulation, and anticancer activity testing. Bioorg Chem 122:105722
Zhao S et al (2021) Ligand-based pharmacophore modeling, virtual screening and biological evaluation to identify novel TGR5 agonists. RSC Adv 11(16):9403–9409. https://doi.org/10.1039/d0ra10168k
Gangwal RP et al (2014) Identification of p38α MAP kinase inhibitors by pharmacophore based virtual screening. J Mol Graph Model 49:18–24. https://doi.org/10.1016/j.jmgm.2014.01.002
Huey R et al (2012) Using AutoDock 4 and AutoDock vina with AutoDockTools: a tutorial. Trends Pharmacol Sci 10550:92037
Rao SN et al (2007) Validation studies of the site-directed docking program LibDock. J Chem Inf Model 47:2159–2171. https://doi.org/10.1021/ci6004299
Wu G et al (2003) Detailed analysis of grid-based molecular docking: a case study of CDOCKER—A CHARMm-based MD docking algorithm. J Comput Chem 24:1549–1562. https://doi.org/10.1002/jcc.10306
Bissantz C et al (2000) Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J Med Chem 43:4759–4767. https://doi.org/10.1021/jm001044l
Liu N et al (2019) Using LeDock as a docking tool for computational drug design. Journal 218:012–143. https://doi.org/10.1088/1755-1315/218/1/012143
Friedman N et al (1997) Bayesian network classifiers. Mach Learn 29:131–163. https://doi.org/10.1023/A:1007465528199
Sugahara S et al (2021) Exact learning augmented naive Bayes classifier. Entropy 23:1703. https://doi.org/10.3390/e23121703
Van Der Spoel D et al (2005) GROMACS: fast, flexible, and free. J Comput chem 26:1701–1718. https://doi.org/10.1002/jcc.20291
Tian C et al (2019) ff19SB: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J Chem Theory Comput 16:528–552. https://doi.org/10.1021/acs.jctc.9b00591
Maier JA et al (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11:3696–3713. https://doi.org/10.1021/acs.jctc.5b00255
Case DA et al (2021) Amber 2021. University of California, San Francisco
Hiscocks J et al (2009) Gaussian 09: IOps reference. In: Caricato M, Frisch MJ (eds). Gaussian
Sousa da Silva AW et al (2012) ACPYPE-Antechamber python parser interface. BMC Res Notes 5:1–8. https://doi.org/10.1186/1756-0500-5-367
McGibbon RT et al (2015) MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys J 109:1528–1532. https://doi.org/10.1016/j.bpj.2015.08.015
Grant BJ et al (2021) The Bio3D packages for structural bioinformatics. Protein Sci 30:20–30. https://doi.org/10.1002/pro.3923
Kumari R et al (2014) g_mmpbsa A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54:1951–1962. https://doi.org/10.1021/ci500020m
Lu S-H et al (2011) The discovery of potential acetylcholinesterase inhibitors: A combination of pharmacophore modeling, virtual screening, and molecular docking studies. J Biomed Sci 18:8. https://doi.org/10.1186/1423-0127-18-8
Lin H-Y et al (2019) Structure-based pharmacophore modeling to discover novel CCR5 inhibitors for HIV-1/cancers therapy. J Biomed Sci 12:10–30. https://doi.org/10.4236/jbise.2019.121002
Weng C-W et al (2022) Hybrid pharmacophore- and structure-based virtual screeningpipeline to identify novel EGFR inhibitors that suppressnon-small cell lung cancer cell growth. Int J Mol Sci 23:34873. https://doi.org/10.3390/ijms23073487
Kohlbacher SM et al (2021) QPHAR: quantitative pharmacophore activity relationship: method and validation. J Cheminformatics 13:57. https://doi.org/10.1186/s13321-021-00537-9
Rogers D et al (2010) Extended-connectivity fingerprints. J Chem Inf Model 50:742–754. https://doi.org/10.1021/ci100050t
Karnati KR et al (2018) Understanding the co-loading and releasing of doxorubicin and paclitaxel using chitosan functionalized single-walled carbon nanotubes by molecular dynamics simulations. Phys Chem Chem Phys 20:9389–9400. https://doi.org/10.1039/C8CP00124C
Acknowledgements
We thank State Key Laboratory of Biotherapy and Cancer Center of West China Hospital for providing the Accelrys DS 3.5 program package to perform this research.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82260693 and 81903543) and Science and Technology Program Project of Gansu Province (20JR5RA534).
Author information
Authors and Affiliations
Contributions
Hui Zhang: conceptualization, methodology, project administration, investigation, supervision, writing—review and editing; Hong-Rui Zhang: methodology, investigation, writing—original draft, investigation, validation; Jian Zhang: validation, resources, formal analysis; Mei-Ling Hu: validation, data curation; Li Ren: data curation, formal analysis; Qing-Qing Luo: validation, data curation; Hua-Zhao Qi: resources, formal analysis.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, H., Zhang, HR., Zhang, J. et al. Discovery of novel S6K1 inhibitors by an ensemble-based virtual screening method and molecular dynamics simulation. J Mol Model 29, 102 (2023). https://doi.org/10.1007/s00894-023-05504-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05504-9