
Abstract
Context
In this discussion, we began building two model, S2O + CHCl•− and O3 + CHCl•−, using DFT-BHandHLYP method, to study their reactions mechanisms on singlet PES. For this purpose, we hope to explore the effects of the difference between sulfur and oxygen atoms on the CHCl•− anion. Experimentalists and computer scientists may utilize the collected data to generate a wide range of hypotheses for experimental phenomena and predictions, allowing them to realize their full potential.
Methods
The ion-molecule reaction mechanism of CHCl•− with S2O and O3 was studied using the DFT-BHandHLYP level of theory with the aug-cc-pVDZ basis set. Our theoretical findings show that Path 6 is the favored reaction pathway for CHCl•− + O3 reaction as identified by the O-abstraction reaction pattern. Comparing to the direct H- and Cl-abstraction mechanisms, the reaction (CHCl•− + S2O) prefers the intramolecular SN2 reaction pattern. Moreover, the calculated results demonstrated that the CHCl•− + S2O reaction is thermodynamically more favorable than the CHCl•− + O3 reaction, which is kinetically more advantageous. As a result, if the required reaction condition in the atmospheric process is met, the O3 reaction will happen more effectively. In terms of kinetics and thermodynamics viewpoints, the CHCl•− anion was very effective in eliminating S2O and O3.
Graphical Abstract








Similar content being viewed by others

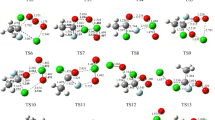
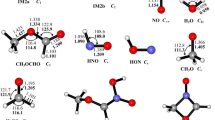
Availability of data and materials
Not applicable.
References
Peiró-Garcı́a J, Nebot-Gil I (2004) An ab initio study on the mechanism of the F + O3 → FO + O2 reaction: comparative reactivity study along the isoelectronic NH2, OH and F radicals series. Chem Phys Lett 391:195–199
Zhang J, Miau TT, Lee YT (1997) Crossed molecular beam study of the reaction Br + O3. J Phys Chem A 101:6922–6930
Kalinowski J, Räsänen M, Gerber RB (2012) Mechanism and electronic transition in the Cl + O3 → ClO + O2 reaction: On the fly dynamics simulations with multi-reference potentials. Chem Phys Lett 535:44–48
Tiwari AJ, Morris JR, Vejerano Jr EP, Hochella MF, Marr LC (2014) Oxidation of C60 aerosols by atmospherically relevant levels of O3. Environ Sci Technol 48:2706–2714
Han Z, Wang J, Zou T, Zhao D, Gao C, Dong J, Pan X (2020) NOx removal from flue gas using an ozone advanced oxidation process with injection of low concentration of ethanol: performance and mechanism. Energy Fuels 34:2080–2088
Yang Y, Liu H (2022) The mechanisms of ozonation for ammonia nitrogen removal: an indirect process. J Environ 10:108525
Machniewski P, Biń A, Kłosek K (2021) Effectiveness of toluene mineralization by gas-phase oxidation over Co(II)/SiO2 catalyst with ozone. Environ 42:3987–3994
Yu H, Ge P, Chen J, Xie H, Luo Y (2017) The degradation mechanism of sulfamethoxazole under ozonation: a DFT study. Environ Sci-Proc Imp 19:379–387
Sun J, Cao H, Zhang S, Li X, He M (2016) Theoretical study on the mechanism of the gas phase reaction of methoxybenzene with ozone. RSC Adv 6:113561–113569
Ataei-Pirkooh A, Alavi A, Kianirad M, Bagherzadeh K, Ghasempour A, Pourdakan O, Adl R, Kiani SJ, Mirzaei M, Mehravi B (2021) Hemispherical asymmetry of the lower stratospheric O3 response to galactic cosmic rays forcing. Sci Rep 11:1–10
Wang N, Wei F, Sun J, Wei B, Mei Q, An Z, Li M, Qiu Z, Bo X, Xie J, Zhan J, He M (2021) Destruction mechanisms of ozone over SARS-CoV-2. Chemosphere 281:130996
Zhao Y, Kuang J, Zhang S, Li X, Wang B, Huang J, Deng S, Wang Y, Yua G (2017) Atmospheric ozonolysis of crotonaldehyde in the absence and presence of hydroxylated silica oligomer cluster adsorption. J Hazard 323:460–470
Kilifarska NA (2017) Ozonation of indomethacin: kinetics, mechanisms and toxicity. ACS Earth Space Chem 1:80–88
Liu J, Li X, Tan Z, Wang W, Yang Y, Zhu Y, Yang S, Song M, Chen S, Wang H, Lu K, Zeng L, Zhang Y (2021) Assessing the ratios of formaldehyde and glyoxal to NO2 as indicators of O3–NOX–VOC sensitivity. Environ Sci Technol 55:10935–10945
Goodarzi M, Vahedpour M, Nazari F (2010) Theoretical study of reaction mechanism for Se + O3. Struct Chem 21:915–922
Kayanuma M, Choe YK, Hagiwara T, Kameda N, Shimoi Y (2021) Theoretical study of the mechanism for the reaction of trimethylaluminum with ozone. ACS Omega 6:26282–26292
Tachikawa H, Abe S (2003) Ozone-water 1: 1 complexes O3-H2O: an ab initio study. Inorg Chem 42:2188–2190
Castle KJ, Black LA, Pedersen TJ (2014) Vibrational relaxation of O3 (ν2) by O (3P). J Phys Chem A 118:4548–4553
Maranzana A, Tonachini G (2020) Multireference study of the H2COO (Criegee Intermediate) + O3 addition: A reaction of possible tropospheric interest. J Phys Chem A 124:1112–1120
Lim S, McArdell CS, von Gunten U (2019) Reactions of aliphatic amines with ozone: kinetics and mechanisms. Water Res 157:514–528
Wang X, Sun J, Han D, Baob L, Meib Q, Wei B, An Z, He M, Yuana S, Xie J, Zhan J, Zhang Q, Wang W (2020) Gaseous and heterogeneous reactions of low-molecular-weight (LMW) unsaturated ketones with O3: mechanisms, kinetics, and effects of mineral dust in tropospheric chemical processes. Chem Eng Sci 395(1):25083
Subbotin OS, Bozhko Y, Zhdanov RK, Gets KV, Belosludov VR, Belosludov RV, Kawazoe Y (2018) Ozone storage capacity in clathrate hydrates formed by O3 + O2 + N2 + CO2 gas mixtures. Phys Chem Phys 20:12637–12641
Hatsugai T, Kiyokawa H, Takeya S, Ohmura R (2021) Improved operation of continuous ozone hydrate production. Chem Eng Technol 44:1677–1685
Wang H, Huang Z, Jiang Z, Jiang Z, Zhang Y, Zhang Z, Shangguan W (2018) Trifunctional C@MnO catalyst for enhanced stable simultaneously catalytic removal of formaldehyde and ozone. ACS Catal 8:3164–3180
Wan X, Wang L, Zhang S, Shi H, Niu J, Wang G, Li W, Chen D, Zhang H, Zhou X, Wang W (2022) Ozone decomposition below room temperature using Mn-based mullite YMn2O5. Environ Sci Technol 56:8746–8755
Liang J, Wei Y, Li Y, Qiang Z, Geng Z (2014) Reaction of CS2 with CHBr•- and CBr2•- in the gas phase: a theoretical mechanistic study. J Iran Chem Soc 11:1345–1352
Feng E, Yu ZJ, Jiang H, Ma X, Nash J (2022) Gas-phase reactivity of phenylcarbyne anions. J Am Chem Soc 144:8576–8590
Zolotov MY, Fegley BJ (1998) Volcanic origin of disulfur monoxide (S2O) on Io. Icarus 133:293–297
Navizet I, Komiha N, Linguerri R, Chambaud G, Rosmus P (2010) On the formation of S2O at low energies: an ab initio study. Chem Phys Lett 500:207–210
Navizet I, Komiha N, Linguerri R, Chambaud G, Rosmus P (2004) Reversible disulfur monoxide (S2O)-forming Retro-Diels-Alder reaction. Disproportionation of S2O to trithio-ozone (S3) and sulfur dioxide (SO2) and reactivities of S2O and S3. J Am Chem Soc 126:9085–9093
Petris G, Troiani A, Angelini G, Ursini O, Bottoni A, Calvaresi M (2007) Isotope exchange in disulfur monoxide-water charged complexes: a mass spectrometric and computational study. J AM SOC MASS SPECTR 18:1664–1671
Sandhiya L, Kolandaivel P, Senthilkumar K (2012) Mechanism and kinetics of the reaction of 1, 4-thioxane with O3 in the atmosphere: a theoretical study. Chem Phys Lett 525:153–159
Liang J, Zhang F, Qi B, Jia W, Liu H, Su Q (2022) Reaction of CHCl•− with HCHO and H2O: A theoretical study. Comput Theor Chem 1217:113932
Goodarzi M, Vahedpour M (2012) Computational study on the multi-channel mechanism of disulfur and ozone reaction. Struct Chem 23:1599–1607
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Jr Montgomery JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. (2003) Gaussian 03, Revision B. 03, Gaussian, Inc., Pittsburgh (PA)
Woon DE, Dunning TH (1993) Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J Chem Phys 98:1358–1371
Mejia-Rodriguez D, Kunitsa A, Apra E, Govind N (2022) Basis set selection for molecular core-level gw calculations. J Chem Theory Comput 18:4919–4926
Filatov M, Lee S, Nakata H, Choi CH (2020) Computation of molecular electron affinities using an ensemble density functional theory method. J Phys Chem A 124:7795–7804
Lee C, Yang W, Parr RG (1988) Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Peng C, Bernhard Schlegel H (1993) Combining synchronous transit and quasi-newton methods to find transition states. Isr J Chem 33:449–454
Hratchian HP, Frisch MJ, Schlegel HB (2010) Steepest descent reaction path integration using a first-order predictor-corrector method. J Chem Phys 133:224101
Tsutsumi T, Ono Y, Arai Z, Taketsugu T (2018) Visualization of the intrinsic reaction coordinate and global reaction route map by classical multidimensional scaling. J Chem Theory Comput 14:4263–4270
Chachiyo T, Chachiyo H (2020) Understanding electron correlation energy through density functional theory. Comput Theor Chem 1172:112669
Pople JA, Head-Gordon M, Raghavachari K (1987) Quadratic configuration interaction. A general technique for determining electron correlation energies. J Chem Phys 87:5968–5975
Ahmadinejad N, Talebi Trai M (2019) Computational NQR−NBO parameters and DFT calculations of ampicillin and zwitterion (monomer and dimer structures). Chem Methodol 3:55–66
Glendening ED, Landis CR, Weinhold F (2012) Pauling’s conceptions of hybridization and resonance in modern quantum chemistry. WIREs Comput Mol Sci 2:1–42
Han H, Suo B, Jiang Z, Wang Y, Wen Z (2008) The potential energy curves of low-lying electronic states of S2O. J Chem Phys 128:184312
Dias N, Gurusinghe RM, Suits AG (2022) Multichannel radical-radical reaction dynamics of NO+ propargyl probed by broadband rotational spectroscopy. J Phys Chem A 126:5354–5362
Liu JY, Long ZW, Mitchell E, Long B (2021) New mechanistic pathways for the reactions of formaldehyde with formic acid catalyzed by sulfuric acid and formaldehyde with sulfuric acid catalyzed by formic acid: formation of potential secondary organic aerosol precursors. ACS Earth Space Chem 5:1363–1372
Peiró-García J, Nebot-Gil I (2002) Ab initio study of the mechanism and thermochemistry of the atmospheric reaction NO + O3 → NO2 + O2. J Phys Chem A 106:10302–10310
Nguyen TH, Nguyen TN, Vu GHT, Nguyen HMT (2022) A quantum chemical investigation of the mechanisms and kinetics of the reactions between methyl radical and n/i-propanol. Comput Theor Chem 1210:113638
Xia W, Zhang R, Xu X, Ma P, Ma C (2022) Spectroscopic features and electronic properties on tetrazole-based energetic cocrystals under external electric field. Comput Theor Chem 1214:113802
Bhujel M, Marshall D, Maccarone A, McKinnon BI, Trevitt A, Silva G, Blanksby S, Poad B (2020) Gas phase reactions of iodide and bromide anions with ozone: evidence for stepwise and reversible reactions. Phys Chem Chem Phys 22:9982–9989
DePuy CH (2002) Understanding organic gas-phase anion molecule reactions. J Org Chem 67:2393–2401
Funding
Project supported: Fundamental Research Funds for the Central Universities (31920220063), the National Natural Science Foundation of China (21968032, 22165025, and 22262031), Chemistry Discipline Innovation Team of Northwest Minzu University (1110130139 and 1110130141).
Author information
Authors and Affiliations
Contributions
Liang Junxi: conceptualization, methodology, and funding acquisition. Zhang Fupeng: validation, formal analysis, and writing original draft. Qi Bomiao: visualization. Lu Mengmeng: funding acquisition and project administration. Pang Shaofeng: data curation. Wang Yanbin: writing review and editing. Su Qiong: project administration.
Corresponding authors
Ethics declarations
Ethics approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fupeng, Z., Junxi, L., Bomiao, Q. et al. A comparable DFT study on reaction of CHCl•− with O3 and S2O. J Mol Model 29, 85 (2023). https://doi.org/10.1007/s00894-023-05483-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05483-x