
Abstract
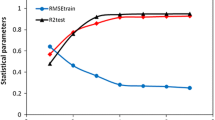
Phenyl acetamide derivatives have a wide range of biological activities, so their research and development can be useful and effective for the design production of new drugs. In this project, quantitative structure–activity relationship (QSAR) was performed. For modeling two methods of multiple linear regression (MLR) and nonlinear regression of support vector machine (SVR) were used. In the MLR stage, the best model with the values of R2train = 0.913 and R2test = 0.881 was selected by stepwise method. In this model, 4 descriptors of BELV2, GATS8p, GATS6e and RDF080m were included, which were used as input for the nonlinear support vector regression method. In the SVR model, the best results were obtained using the radial Gaussian kernel function (RBF) with R2train = 0.978 and R2test = 0.990. In the next step, using molecular docking and molecular dynamic simulation methods, the interaction between phenyl acetamide derivatives and the sirtuin 2 protein was investigated. Examining the results of molecular docking, it was observed that these derivatives formed complexes by forming hydrogen and hydrophobic bonds with the sirtuin 2 protein. Also, the results of molecular dynamic simulation show that phenyl acetamide compounds form stable complex with the sirtuin 2 protein, and it was found that the compounds with more activity have formed a number of hydrogen bonds with the protein.










Similar content being viewed by others
Data availability
The data that support the findings of this study are available within the article.
Code availability
Not applicable.
References
Seyfried TN, Flores RE, Poff AM, D’Agostino DP (2014) Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis. https://doi.org/10.1093/carcin/bgt480
Seyfried TN, Shelton LM (2010) Cancer as a metabolic disease. Nutr Metab. https://doi.org/10.1186/1743-7075-7-7
Mei Zh, Zhang X, Yi J, Huang J, He J, Tao Y (2016) Sirtuins in metabolism, DNA repair, and cancer. J Exp Clin Cancer Res. https://doi.org/10.1186/s13046-016-0461-5
Hoffmann G, Breitenbücher F, Schuler M, Ehrenhofer-Murray AE (2014) A Novel Sirtuin 2 (SIRT2) Inhibitor with p53-dependent Pro-apoptotic Activity in Non-small Cell Lung Cancer. J Biol Chem. https://doi.org/10.1074/jbc.M113.487736
Lee IH (2019) Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Exp Mol Med. https://doi.org/10.1038/s12276-019-0302-7
Bosch-Presegué L, Vaquero A (2011) The Dual Role of Sirtuins in cancer. SAGE. https://doi.org/10.1177/1947601911417862
Gomes P, Fleming Outeiro T, Cavadas C (2015) Emerging Role of Sirtuin 2 in the Regulation of Mammalian Metabolism. Trends Pharmacol Sci. https://doi.org/10.1016/j.tips.2015.08.001
Yang L, Xi Ma, Yuan Ch, He Y, Li L, Fang S, Xia W, He T, Qian S, Xu Zh, Li G, Wang Zh (2017) Discovery of 2-((dimethylpyrimidin-2-yl) thio)-N- phenylacetamide derivatives as new potent and selective human sirtuin 2 inhibitors. Eur J Med Chem. https://doi.org/10.1016/j.ejmech.2017.04.010
Aki-Sener E, Bingol KK, Temiz-Arpaci O, Yalcin I, Altanlar N (2002) Synthesis and microbiological activity of some N-(2-hydroxy-4-substitutedphennyl) benzamides, phenyl acetamides and furamides as the possible metabolites of antimicrobial active benzoxazoles. IL FARMACO. https://doi.org/10.1016/S0014-827X(02)01226-0
SahuN P, Pal Ch, Mandal NB, Banerjee S, Raha M, Kundu AP, Basu A, Ghosh M, Roy K, Bandyopadhyay S (2002) Synthesis of a Novel Quinoline Derivatives, 2-(2-Methylquinolin-4-ylamino)-N-phenylacetamide—A potential Antileishmanial agent. Bioorg Med Chem. https://doi.org/10.1016/S0968-0896(02)00046-9
Soyer Z, Kilic FS, Erol K, Pabuccuoglu V (2004) Synthesis and anticonvulsant activity of some ?-(1H-imidazol-1-yl)-N-phenylacetamide and propionamide derivatives. ILFARMACO. https://doi.org/10.1016/j.farmac.2003.07.011
Ertan T, Yildiz I, Ozkan S, Temiz-Arpaci O, Kaynak F, Yalcin I, Aki-Sener E, Abbasoglu U (2007) Synthesis and biological evaluation of new N-(2-hydroxy-4(or 5)- nitro/aminophenyl)benzamides and phenylacetamides as antimicrobial agents. Bioorg Med Chem. https://doi.org/10.1016/J.BMC.2006.12.035
Bu M, Cao T, Li H, Guo M, Yang BB, Zeng Ch, Zhou Y, Zhang N, Hu L (2017) Synthesis and biological evaluation of novel steroidal 5a,8a-epidioxyandrost-6-ene-3ß-ol-17-(O-phenylacetamide)oxime derivatives as potential anticancer agents. Bioorg Med Chem Lett. https://doi.org/10.1016/j.bmcl.2017.06.048
Farshad S, Darvish Ganji M (2020) Theoretical study of interaction between aspirine drug and Al-soped graphene nanostructure toward designing of suitable nanocarrier for drug delivery. Medical Sciences. https://doi.org/10.29252/iau.30.2.141
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools 4:automated docking with selective receptor flexibility. J Comput Chem. https://doi.org/10.1002/jcc.21256
P. Norgan A, Coffman PK, Kocher JPA, Katzmann DPJ, Sosa C (2011) Multilevel parallelization of Autodock 4.2. J Cheminform. https://doi.org/10.1186/1758-2946-3-12
Safarizadeh H, Garkani-Nejad Z (2019) Molecular docking, Molecular dynamics simulations and QSAR studies on some of 2-arylethenylquinoline derivatives for inhibition of Alzheime’s amyloid-beta aggregation: Insight into mechanism of interactions and parameters for design of new inhibitors. J Mol Graph Model. https://doi.org/10.1016/j.jmgm.2018.11.019
Mortier J, Rakers C, Bermudez MM, Murgueitio MS, Riniker S, Wolber G (2015) The impact of molecular dynamics on drug design: applications for the characterization of ligand-macromolecule complexes. Drug Discov Today. https://doi.org/10.1016/j.drudis.2015.01.003
Durrant JD, McCammon JA (2011) Molecular dynamics simulations and drug discovery. J Biol. https://doi.org/10.1186/1741-7007-9-71
Pronk S, Páll S, SchulzR, Larsson P, Bjelkmar P, Apostolov R, R. Shirts M, C. Smith J, M. Kasson P, Van der Spoel D, Hess B, Lindahl E (2013) GROMACS 4.5: a high throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. https://doi.org/10.1093/bioinformatics/btt055
Lins RD, Hünenberger PhH (2005) A new GROMOS force field for hexopyranose-based carbohydrates. J Comput Chem. https://doi.org/10.1002/jcc.20275
Van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark E, A, J. C. Berendsen H, (2005) GROMACS:Fast, flexible, and free. J Comput Chem. https://doi.org/10.1002/jcc.20291
SafarizadehH G-N (2019) Investigation of MI-2 analogues as MALT1 inhibitors to treat of diffuse large B-Cell lymphoma through combined molecular dynamics simulation, molecular docking and QSAR techniques and design of new inhibitors. J Mol Struct. https://doi.org/10.1016/j.molstruc.2018.12.022
Chen S, Wang H, Zhang J, Lu S, Xiang Y (2020) Effect of side chain on the electrochemical performance of poly (ether ether ketone) based anion-exchange membrane: A molecular dynamics study. J Membr Sci. https://doi.org/10.1016/j.memsci.2020.118105
Xi L, Wang Y, He Q, Zhang Q, Du L (2016) Interaction between pin 1 and its natural product inhibitor epigallocatechin-3- gallate by spectroscopy and molecular dynamics simulations. Spectrochim Acta. https://doi.org/10.1016/j.saa.2016.06.036
Onufriev A, Bashford D, Case AD (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. https://doi.org/10.1002/PROT.20033
Humphrey W, DalkeA SK (1995) VMD: visual molecular dynamics. J Mol Graph. https://doi.org/10.1016/0263-7855(96)00018-5
Pettersen FED, Goddard DT, Huang CC, CouchGreenblatt SGMD, Meng CE, Ferrin ET (2004) UCSF Chimera- A visualization system for exploratory research and analysis. J Comput Chem. https://doi.org/10.1002/jcc.20084
Kubinyi H (1997) QSAR and 3D QSAR in drug design part 1: methodology. Drug Discov Today. https://doi.org/10.1016/S1359-6446(97)01079-9
Einax WJ (2008) Chemometrics in analytical chemistry. Anal Bioanal Chem. https://doi.org/10.1007/s00216-007-1786-x
Shafieyoon P, Mehdipour E, Mary Y.S (2019) Synthesis, characterization and biological investigation of glycine-based sulfonamide derivatives and its complex: Vibration assignment, HOMO-LUMO analysis, MEP and molecular docking. J Mol Struct. https://doi.org/10.1016/j.molstruc.2018.12.067
Kumer A, Sarker N, Paul S, Zannat A (2019) The Theoretical Prediction of Thermophysical properties, HOMO, LUMO, QSAR and Biological Indics of Cannabinoids (CBD) and Tetrahhdrocannabinol (THC) by Computational Chemistry. Adv J Chem A. https://doi.org/10.33945/SAMI/AJCA.2019.2.190202
Mouri A, Consonni V, Pavan M, Todeschini R (2006) DRAGON SOFTWARE: ANEASY APPROACH TO MOLECULAR DESCRIPTOR CALCULATIONS. MATCH Commun Math Comput Chem 56:237–248
Gharagheizi F (2008) Quantitative structure-property relationship for prediction of the lower flammability limit of pure compounds. Energy Fuels. https://doi.org/10.1021/ef800375b
Luu QH, Lau MF, Ng PHS, Yueh Chen T (2021) Testing multiple linear regression systems with metamorphic testing. J Syst Softw. https://doi.org/10.1016/j.jss.2021.111062
Preacher JK, Curran JP, Bauer JD (2006) Computational tools for probing interactions in multiple linear regression, multilevel modeling, and latent curve analysis. J Educ Behav Stat. https://doi.org/10.3102/10769986031004437
Zhi-qiang J, Han-guang F, Ling-jun L (2005) Support Vector Machine for mechanical faults classification. J Zhejiang Univ SCI. https://doi.org/10.1631/jzus.2005.A0433
Smola JA, Schölkopf B (2004) A tutorial on support vector regression. Stat Comput. https://doi.org/10.1023/B:STCO.0000035301.49549.88
Üstün B, Melssen WJ, Oudenhuijzen M, Buydens LMC (2005) Determination of Optimal support vector Regression parameters by Genetic Algorithms and Simplex Optimization. Anal Chim Acta. https://doi.org/10.1016/j.aca.2004.12.024
M. Balabin R, I. Lomakina E (2011) Support vector machine regression (SVR/LS-SVM)- an alternative to neural networks (ANN) for analytical chemistry? Comparison of nonlinear methods on near infrared (NIR) spectroscopy data. Analyst. https://doi.org/10.1039/c0an00387e
Sánches VD (2003) Advanced Support Vector Machines and kernel methods. Neurocomputing. https://doi.org/10.1016/S0925-2312(03)00373-4
Yu L, Yau X, Wang S, Lai KK (2011) Credit risk evaluation using a weighted least squares SVM classifier with design of experiment for parameter selection. Expert Syst Appl. https://doi.org/10.1016/j.eswa.2011.06.023
Todeschini R, Cosonni V (2000) Handbook of Molecular Descriptors. Weinheim. New York. Chichester. Brisbane Singapore Toronto.
Asadollahi-Baboli M, Mani-Varnosfaderani A (2015) Therapeutic index modeling and predictive QSAR of novel thiazolidin-4-one analogs against Toxoplasma gondii. Eur J Pharm Sci. https://doi.org/10.1016/j.ejps.2015.01.014
Mahmud A. W, Shallangwa G. A, Uzairu A (2019) Quantitative structure –activity relationships (QSAR) study on novel 4-amidinoquinoline and 10-amidinobenzonaphthyridine derivatives as potent antimalaria agent. The journal of engineering and exact sciences. https://doi.org/10.18540/jcecvl5iss3pp0271-0282
Pourbasheer E, Aalizadeh R, Ganjali MR (2019) QSAR study of CK2 inhibitors by GA-MLR and GA-SVM methods. Arab J Chem. https://doi.org/10.1016/j.arabjc.2014.12.021
Kumar Gupta A, A. Gupta R, Kumar Soni L, Kaskhedikar S. G, (2008) Exploration of physicochemical properties and molecular modeling studies of 2-sulfonyl-phenyl-3-phenyl-indole analogs as cyclooxygenase-2 inhibitors. Eur J Med Chem. https://doi.org/10.1016/j.ejmech.2007.06.022
Wang T, Tang L, Luan F, D. S. Cordeiro M. N, (2018) Prediction of the toxicity of binary mixtures by QSAR approach using the hypothetical descriptors. Int J Mol Sci. https://doi.org/10.3390/ijms19113423
Riniker S, P. Eichenberger A, F. van Gunsteren W, (2012) Solvating atomic level fine-grained proteins in supra-molecular level coarse-grained water for molecular dynamics simulations. Eur Biophys J. https://doi.org/10.1007/s00249-012-0837-1
Skariyachan S, Khangwal I, Niranjan V, Kango N, Shukla P (2020) Deciphering effectual binding potential of xylo-substrates towards xylose isomerase and xylokinase through molecular docking and molecular dynamic simulation. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1772882
Acknowledgements
The authors gratefully acknowledge the Shahid Bahonar University of Kerman.
Author information
Authors and Affiliations
Contributions
S. I. contributed to QSAR analysis, molecular docking and molecular dynamic simulation analysis, and writing the first draft of the manuscript. Z. G. contributed to project administration, supervision, methodology, interpretation of the results, reviewing, and editing.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ilaghi-Hoseini, S., Garkani-Nejad, Z. Research and study of 2-((4,6 dimethyl pyrimidine-2-yle) thio)-N-phenyl acetamide derivatives as inhibitors of sirtuin 2 protein for the treatment of cancer using QSAR, molecular docking and molecular dynamic simulation. J Mol Model 28, 343 (2022). https://doi.org/10.1007/s00894-022-05288-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05288-4