
Abstract
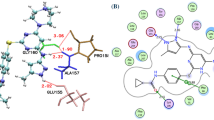
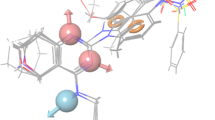
Human phosphatidylethanolamine binding protein 1 (hPEBP1) is a novel target affecting many cellular signaling pathways involved in the formation of metastases. It can be used in the treatment of many cases of cancer. For these reasons, pharmaceutical companies use computational approaches, including multi-QSAR (2D, 3D, and hologram QSAR) analysis, homology modeling, molecular docking analysis, and molecular dynamic simulations, to speed up the drug discovery process. In this paper, QSAR modeling was conducted using two quantum chemistry optimization methods (AM1 and DFT levels). As per PLS results, we found that the DFT/B3LYP method presents high predictability according to 2D-QSAR, CoMFA, CoMSIA, and hologram QSAR studies, with Q2 of 0.81, 0.67, 0.79, and 0.67, and external power with R2pred of 0.78, 0.58, 0.66, and 0.56, respectively. This result has been validated by CoMFA/CoMSIA graphics, which suggests that electrostatic fields combined with hydrogen bond donor/acceptor fields are beneficial to the antiproliferative activity. While the hologram QSAR models show the contributions of each fragment in improving the activity. The results from QSAR analyses revealed that ursolic acids with heterocyclic rings could improve the activities. Ramachandran plot validated the modeled PEBP1 protein. Molecular docking and MD simulations revealed that the hydrophobic and hydrogen bond interactions are dominant in the PEBP1’s pocket. These results were used to predict in silico structures of three new compounds with potential anticancer activity. Similar molecular docking stability studies and molecular dynamics simulations were conducted.
Graphical abstract



















Similar content being viewed by others

Data availability
The data is integrated into the manuscript.
Code availability
Not applicable.
Abbreviations
- hPEBP1:
-
Human phosphatidylethanolamine binding protein 1
- QSARs:
-
Quantitative structure–activity relationships
- 2D:
-
Two dimensional
- 3D:
-
Three dimensional
- DFT:
-
Density functional theory
- AM1:
-
Austin model 1
- MDs:
-
Molecular dynamics
- CoMFA:
-
Comparative molecular field analysis
- CoMSIA:
-
Comparative molecular similarity indices analysis
- HQSAR:
-
Hologram quantitative structure–activity relationships
- GMQE score:
-
Global Model Quality Estimation
- OECD:
-
Organization for Economic Co-operation and Development
References
Bray F, Ferlay J, Soerjomataram I et al (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J Clinicians 68:394–424. https://doi.org/10.3322/caac.21492
Schoentgen F, Jonic S (2018) PEBP1/RKIP: from multiple functions to a common role in cellular processes. arXiv preprint arXiv:1802.02378
Beshir AB, Ren G, Magpusao AN et al (2010) Raf kinase inhibitor protein suppresses nuclear factor-κB-dependent cancer cell invasion through negative regulation of matrix metalloproteinase expression. Cancer Lett 299:137–149. https://doi.org/10.1016/j.canlet.2010.08.012
Al-Mulla F, Bitar MS, Al-Maghrebi M et al (2011) Raf kinase inhibitor protein RKIP enhances signaling by glycogen synthase kinase-3β. Cancer Res 71:1334–1343. https://doi.org/10.1158/0008-5472.CAN-10-3102
Rath O, Park S, Tang H et al (2008) The RKIP (Raf-1 kinase inhibitor protein) conserved pocket binds to the phosphorylated N-region of Raf-1 and inhibits the Raf-1-mediated activated phosphorylation of MEK. Cell Signal 20:935–941. https://doi.org/10.1016/j.cellsig.2008.01.012
Yan X-F, Xiao H-M, Gong X-D, Ju X-H (2006) A comparison of semiempirical and first principle methods for establishing toxicological QSARs of nitroaromatics. J Mol Struct (Thoechem) 764:141–148. https://doi.org/10.1016/j.theochem.2006.02.018
Puzyn T, Leszczynski J, Cronin MTD (2010) Recent advances in QSAR studies: methods and applications. Challenges and advances in computational chemistry and physics. Springer, Dordrecht, New York
Cronin MTD, Livingstone D (2004) Predicting chemical toxicity and fate. CRC Press
Kubinyi H (1998) Comparative Molecular Field Analysis (CoMFA). Encyclopedia Comput Chem 1:448–460
Ghemtio L, Zhang Y, Xhaard H (2012) CoMFA/CoMSIA and pharmacophore modeling as a powerful tools for efficient virtual screening: application to anti-leishmanial betulin derivatives. In: Virtual Screening, 55–82
Lowis DR (1997) HQSAR: a new, highly predictive QSAR technique. Tripos Tech Notes 1:17
Muhammed MT, Aki-Yalcin E (2019) Homology modeling in drug discovery: overview, current applications, and future perspectives. Chem Biol Drug Des 93:12–20. https://doi.org/10.1111/cbdd.13388
Safarizadeh H, Garkani-Nejad Z (2019) Molecular docking, molecular dynamics simulations and QSAR studies on some of 2-arylethenylquinoline derivatives for inhibition of Alzheimer’s amyloid-beta aggregation: insight into mechanism of interactions and parameters for design of new inhibitors. J Mol Graph Model 87:129–143. https://doi.org/10.1016/j.jmgm.2018.11.019
Muneeswaran G, Pandiaraj M, Kartheeswaran S et al (2018) Molecular dynamics simulation approach to explore atomistic molecular mechanism of peroxidase activity of apoptotic cytochrome c mutants. Informatics Med Unlocked 11:51–60. https://doi.org/10.1016/j.imu.2018.04.003
Woźniak Ł, Skąpska S, Marszałek K (2015) Ursolic acid—a pentacyclic triterpenoid with a wide spectrum of pharmacological activities. Molecules 20:20614–20641. https://doi.org/10.3390/molecules201119721
Mourya A, Akhtar A, Ahuja S et al (2018) Synergistic action of ursolic acid and metformin in experimental model of insulin resistance and related behavioral alterations. Eur J Pharmacol 835:31–40. https://doi.org/10.1016/j.ejphar.2018.07.056
López-Hortas L, Pérez-Larrán P, González-Muñoz MJ et al (2018) Recent developments on the extraction and application of ursolic acid. A review. Food Res Int 103:130–149. https://doi.org/10.1016/j.foodres.2017.10.028
Hua S-X, Huang R-Z, Ye M-Y et al (2015) Design, synthesis and in vitro evaluation of novel ursolic acid derivatives as potential anticancer agents. Eur J Med Chem 95:435–452. https://doi.org/10.1016/j.ejmech.2015.03.051
Tang Q, Liu Y, Li T et al (2016) A novel co-drug of aspirin and ursolic acid interrupts adhesion, invasion and migration of cancer cells to vascular endothelium via regulating EMT and EGFR-mediated signaling pathways: multiple targets for cancer metastasis prevention and treatment. Oncotarget 7. https://doi.org/10.18632/oncotarget.12232
Mlala S, Oyedeji AO, Gondwe M, Oyedeji OO (2019) Ursolic acid and its derivatives as bioactive agents. Molecules 24:2751. https://doi.org/10.3390/molecules24152751
Qian Z, Wang X, Song Z et al (2015) A phase I trial to evaluate the multiple-dose safety and antitumor activity of ursolic acid liposomes in subjects with advanced solid tumors. Biomed Res Int 2015:1–7. https://doi.org/10.1155/2015/809714
Sultana N (2011) Clinically useful anticancer, antitumor, and antiwrinkle agent, ursolic acid and related derivatives as medicinally important natural product. J Enzyme Inhib Med Chem 26:616–642. https://doi.org/10.3109/14756366.2010.546793
Iqbal J, Abbasi BA, Ahmad R et al (2018) Ursolic acid a promising candidate in the therapeutics of breast cancer: current status and future implications. Biomed Pharmacother 108:752–756. https://doi.org/10.1016/j.biopha.2018.09.096
Leal AS, Wang R, Salvador JAR, Jing Y (2012) Synthesis of novel ursolic acid heterocyclic derivatives with improved abilities of antiproliferation and induction of p53, p21waf1 and NOXA in pancreatic cancer cells. Bioorg Med Chem 20:5774–5786. https://doi.org/10.1016/j.bmc.2012.08.010
Gaussian 09 (2009) R.A.: 1, mj frisch, gw trucks, hb schlegel, ge scuseria, ma robb, jr cheeseman, g. Scalmani, v. Barone, b. Mennucci, ga petersson et al., gaussian. Inc Wallingford CT. 121, 150
Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140:A1133–A1138. https://doi.org/10.1103/PhysRev.140.A1133
Lazrak M, Toufik H, Bouzzine SM et al (2018) The computational study of bridge effect in D-π-A photosensitive dyes, based on triphenylamine. IOP Conf Ser Earth Environ Sci 161:012021. https://doi.org/10.1088/1755-1315/161/1/012021
Lazrak M, Toufik H, Bouzzine SM, Lamchouri F (2020) Bridge effect on the charge transfer and optoelectronic properties of triphenylamine-based organic dye sensitized solar cells: theoretical approach. Res Chem Intermed 46:3961–3978. https://doi.org/10.1007/s11164-020-04184-x
Casida ME (1995) Time-dependent density functional response theory for molecules. In: Recent Advances in Computational Chemistry. WORLD SCIENTIFIC 155–192. https://doi.org/10.1142/9789812830586_0005
Ennehary S, Toufik H, Bouzzine SM, Lamchouri F (2020) Effect of the alkyl chain length on the optoelectronic properties of organic dyes: theoretical approach. J Comput Electron 19:840–848. https://doi.org/10.1007/s10825-020-01486-6
Gramatica P, Chirico N, Papa E et al (2013) QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J Comput Chem 34:2121–2132. https://doi.org/10.1002/jcc.23361
Stitou M, Toufik H, Akabli T et al (2018) 2D-QSAR method of lupane-type saponins the treatement of cancer cell line. RHAZES: Green Appl Chem 2:33–45
Akabli T, Toufik H, Yasri A et al (2018) Combining ligand-based and structure-based drug design approaches to study the structure-activity relationships of a β-carboline derivative series. Struct Chem 29:1637–1645. https://doi.org/10.1007/s11224-018-1141-1
O’brien RM, (2007) A caution regarding rules of thumb for variance inflation factors. Qual Quant 41:673–690. https://doi.org/10.1007/s11135-006-9018-6
Golbraikh A, Tropsha A (2000) Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. Mol Divers. 231-243. https://doi.org/10.1023/A:1021372108686
ReenuVikas, (2016) Evaluating the role of electron-correlation in the external prediction of the toxicity of nitrobenzenes towards Tetrahymena pyriformis. New J Chem 40:2343–2353. https://doi.org/10.1039/C5NJ02552D
Stitou M, Toufik H, Bouachrine M et al (2019) Machine learning algorithms used in quantitative structure-activity relationships studies as new approaches in drug discovery. In: 2019 International Conference on Intelligent Systems and Advanced Computing Sciences (ISACS), IEEE, Taza, Morocco, 1–8. https://doi.org/10.1109/ISACS48493.2019.9068917
Rücker C, Rücker G, Meringer M (2007) y-randomization and its variants in QSPR/QSAR. J Chem Inf Model 47:2345–2357. https://doi.org/10.1021/ci700157b
Roy K, Kar S, Ambure P (2015) On a simple approach for determining applicability domain of QSAR models. Chemom Intell Lab Syst 145:22–29. https://doi.org/10.1016/j.chemolab.2015.04.013
Pourbasheer E, Aalizadeh R, Shokouhi Tabar S et al (2014) 2D and 3D quantitative structure–activity relationship study of hepatitis C virus NS5B polymerase inhibitors by comparative molecular field analysis and comparative molecular similarity indices analysis methods. J Chem Inf Model 54:2902–2914. https://doi.org/10.1021/ci500216c
Hadni H, Elhallaoui M (2020) 2D and 3D-QSAR, molecular docking and ADMET properties in silico studies of azaaurones as antimalarial agents. New J Chem 44:6553–6565. https://doi.org/10.1039/C9NJ05767F
Halim SA, Zaheer-ul-Haq, (2015) Structure based 3D-QSAR studies of interleukin-2 inhibitors: comparing the quality and predictivity of 3D-QSAR models obtained from different alignment methods and charge calculations. Chem Biol Interact 238:9–24. https://doi.org/10.1016/j.cbi.2015.05.018
SYBYL/QSAR. Molecular Modelling Software, Tripos Inc., 1699 S. Hanley Road, St. Louis, MO 63944, USA
Stitou M, Toufik H, Bouachrine M, Lamchouri F (2020) Quantitative structure–activity relationships analysis, homology modeling, docking and molecular dynamics studies of triterpenoid saponins as Kirsten rat sarcoma inhibitors. J Biomol Struct Dyn 39:152–170. https://doi.org/10.1080/07391102.2019.1707122
Matysiak J, Niewiadomy A (2017) QSAR models of antiproliferative activity of imidazo[2,1-b][1,3,4]thiadiazoles in various cancer cell lines. Mol Divers 21:211–218. https://doi.org/10.1007/s11030-016-9705-8
Wang X, Yan J, Wang M et al (2018) Synthesis and three-dimensional quantitative structure-activity relationship study of quinazoline derivatives containing a 1,3,4-oxadiazole moiety as efficient inhibitors against Xanthomonas axonopodis pv. citri. Mol Divers 22:791–802. https://doi.org/10.1007/s11030-018-9837-0
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. https://doi.org/10.1093/bioinformatics/bti770
Ye W-L, Zhang L-X, Guan Y-D et al (2019) Virtual screening and experimental validation of eEF2K inhibitors by combining homology modeling, QSAR and molecular docking from FDA approved drugs. New J Chem 43:19097–19106. https://doi.org/10.1039/C9NJ02600B
Benkert P, Tosatto SCE, Schomburg D (2008) QMEAN: A comprehensive scoring function for model quality assessment. Proteins Struct Funct Bioinformatics 71:261–277. https://doi.org/10.1002/prot.21715
Van Der Spoel D, Lindahl E, Hess B et al (2005) GROMACS: Fast, flexible, and free. J Comput Chem 26:1701–1718. https://doi.org/10.1002/jcc.20291
Vanommeslaeghe K, Hatcher E, Acharya C et al (2009) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem NA-NA. https://doi.org/10.1002/jcc.21367
Huang J, MacKerell AD (2013) CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 34:2135–2145. https://doi.org/10.1002/jcc.23354
Jorgensen WL, Tirado-Rives J The OPLS potential functions for proteins. Energy minimizations for crystals of cyclic peptides and crambin. J Am Chem Soc 110:1657–1666. https://doi.org/10.1021/ja00214a001
Berendsen HJC, Postma JPM, van Gunsteren WF et al (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690. https://doi.org/10.1063/1.448118
Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7190. https://doi.org/10.1063/1.328693
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092. https://doi.org/10.1063/1.464397
Ryckaert J-P, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341. https://doi.org/10.1016/0021-9991(77)90098-5
Walton IM, Cox JM, Benson CA et al (2016) The role of atropisomers on the photo-reactivity and fatigue of diarylethene-based metal–organic frameworks. New J Chem 40:101–106. https://doi.org/10.1039/C5NJ01718A
Frimand K, Jalkanen KJ (2002) SCC-TB, DFT/B3LYP, MP2, AM1, PM3 and RHF study of ethylene oxide and propylene oxide structures, VA and VCD spectra. Chem Phys 279:161–178. https://doi.org/10.1016/S0301-0104(02)00457-3
Pasha FA, Srivastava HK, Singh PP (2005) Comparative QSAR study of phenol derivatives with the help of density functional theory. Bioorg Med Chem 13:6823–6829. https://doi.org/10.1016/j.bmc.2005.07.064
Deeb O, Clare BW (2008) Comparison of AM1 and B3LYP-DFT for inhibition of MAO-A by phenylisopropylamines: a QSAR study. Chem Biol Drug Design 71:352–362. https://doi.org/10.1111/j.1747-0285.2008.00643.x
Myint KZ, Xie X-Q (2010) Recent advances in fragment-based QSAR and multi-dimensional QSAR methods. IJMS 11:3846–3866. https://doi.org/10.3390/ijms11103846
Rost B, Sander C (1996) Bridging the protein sequence-structure gap by structure predictions. Annu Rev Biophys Biomol Struct 25:113–136
Weiner PK, Langridge R, Blaney JM et al (1982) Electrostatic potential molecular surfaces. Proc Natl Acad Sci 79:3754–3758. https://doi.org/10.1073/pnas.79.12.3754
Rathi PC, Ludlow RF, Verdonk ML (2019) Practical high-quality electrostatic potential surfaces for drug discovery using a graph-convolutional deep neural network. J Med Chem 63:8778–8790. https://doi.org/10.1021/acs.jmedchem.9b01129
Monajjemil M, Oliaey AR (2009) Gyration radius and energy study at different temperatures for acetylcholine receptor protein in gas phase by Monte Carlo, molecular and Langevin dynamics simulations. J Phys Theor Chem Islamic Azad Univ Iran 5:195–201
Funding
This work was supported by the National Center for Scientific and Technical Research (CNRST — Morocco) as part of the Research Excellence Awards Program (no. 34USMBA2017).
Author information
Authors and Affiliations
Contributions
All persons who meet authorship criteria are listed as authors, and all authors certify that they have participated sufficiently in the work to take public responsibility for the content, including participation in the concept, design, analysis, writing, or revision of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Stitou, M., Toufik, H., Akabli, T. et al. Virtual screening of PEBP1 inhibitors by combining 2D/3D-QSAR analysis, hologram QSAR, homology modeling, molecular docking analysis, and molecular dynamic simulations. J Mol Model 28, 145 (2022). https://doi.org/10.1007/s00894-022-05143-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05143-6