
Abstract
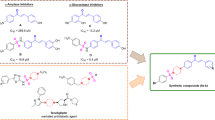
In the present study, a quantitative relationship between the biological inhibitory activity of alpha-amylase and molecular structures of novel benzimidazole derivatives is analyzed in silico. The best QSAR model screened via MLR technique indicated that the exact mass, topological diameter and numerical rotational bonding structural properties of benzimidazole derivatives highly affect the bioactivity of these compounds against α-amylase. Based on the structural properties identified via linear QSAR model favorable for improving pIC50 of benzimidazole derivatives, fourteen new molecules bearing benzimidazole radicals were designed and their biological inhibitory activity against α-amylase was improved. QSAR model predictions showed that the designed molecules exhibited a higher potential biological level activity IC50 than acarbose used in positive control (IC50= 1.46 μM). Screening of drug-like properties, pharmacokinetics and toxicity of the proposed molecules led to select three molecules as candidates for use as drug aid to ingest starch and glycogen. As a result, using molecular docking simulations, the docking poses of the three molecules inside the α-amylase receptor pocket (PDB code: 1HNY) were predicted. Also, the most important potential interactions between the active amino acid sites in α-amylase protein pocket and the proposed drug molecules were described. The obtained hypotheses regarding the stability of the proposed molecules inside α-amylase pocket were validated by carrying out molecular dynamic simulations in aqueous background similar to the ones of proteins. The DM results confirmed the optimal stability of the α-amylase backbone with the drug molecules proposed in this computational investigation.
Graphical abstract









Similar content being viewed by others

Data Availability
Not applicable.
Code availability
Not applicable.
References
Danaei G, Lawes CM, Vander Hoorn S, Murray CJ, Ezzati M (2006) Global and regional mortality from ischaemic heart disease and stroke attributable to higher-than-optimum blood glucose concentration: comparative risk assessment. Lancet 368:1651–1659
The effects of diabetes on your body, Healthline (2014) https://www.healthline.com/health/diabetes/effects-on-body. Accessed 26 Mar 2022
Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, Di Angelantonio E, Ingelsson E, Lawlor DA, Selvin E, Stampfer M (2010) Emerging risk factors collaboration diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet 375:2215–2222
Chun BY, Rizzo JF III (2016) Dominant optic atrophy: updates on the pathophysiology and clinical manifestations of the optic atrophy 1 mutation. Curr Opin Ophthalmol 27:475–480
Saran R, Robinson B, Abbott KC, Agodoa LY, Albertus P, Ayanian J, Balkrishnan R, Bragg-Gresham J, Cao J, Chen JL (2017) US renal data system 2016 annual data report: epidemiology of kidney disease in the United States. Am J Kidney Dis 69:A7–A8
Diabetes mellitus guide: causes, symptoms and treatment options (n.d.) https://www.drugs.com/health-guide/diabetes-mellitus.html. Accessed 26 Mar 2022
Funke I, Melzig MF (2006) Plantas tradicionalmente utilizadas na terapia da diabetes: fitomedicamentos como inibidores da atividade alfa-amilase, Revista Brasileira de. Farmacognosia 16:1–5
Sales PM, Souza PM, Simeoni LA, Magalhães PO, Silveira D (2012) α-Amylase inhibitors: a review of raw material and isolated compounds from plant source. J Pharm Pharm Sci 15:141–183
Van De Laar FA, Lucassen PL, Akkermans RP, Van De Lisdonk EH, Rutten GE, Van Weel C (2005) α-Glucosidase inhibitors for patients with type 2 diabetes: results from a Cochrane systematic review and meta-analysis. Diabetes Care 28:154–163
Arshad T, Khan KM, Rasool N, Salar U, Hussain S, Tahir T, Ashraf M, Waddod A, Riaz M, Perveen S, Taha M, Ismail NH (2016) Syntheses, in vitro evaluation and molecular docking studies of 5-bromo-2-aryl benzimidazoles as α-glucosidase inhibitors. Med Chem Res 25:2058–2069. https://doi.org/10.1007/s00044-016-1614-y
Aminpour M, Montemagno C, Tuszynski JA (2019) An overview of molecular modeling for drug discovery with specific illustrative examples of applications. Molecules 24:1693
Chalkha M, Akhazzane M, Moussaid FZ, Daoui O, Nakkabi A, Bakhouch M, Chtita S, Elkhattabi S, Housseini AI, Yazidi ME (2022) Design, synthesis, characterization, in vitro screening, molecular docking, 3D-QSAR, and ADME-Tox investigations of novel pyrazole derivatives as antimicrobial agents. New J Chem. 46:2747–2760 https://doi.org/10.1039/D1NJ05621B
Adegboye AA, Khan KM, Salar U, Aboaba SA, Chigurupati S, Fatima I, Taha M, Wadood A, Mohammad JI, Khan H (2018) 2-Aryl benzimidazoles: synthesis, in vitro α-amylase inhibitory activity, and molecular docking study. Eur J Med Chem 150:248–260
ChemOffice, PerkinElmer Informatics.http://www.cambridgesoft.com, 2016. Accessed 26 Mar 2022
MarvinSketch 5.11.4., Chem Axon 2012. https://chemaxon.com/products/marvin. Accessed 26 Mar 2022
ACDLABS 10. Advanced Chemistry Development. Inc. Toronto. ON. Canada 2015. Available:(http://www.acdlabs.com/ressources/freeware/chemsketch/). Accessed 26 Mar 2022
XLSTAT, Software, XLSTAT company.www.xlstat.com, 2013. Accessed 26 Mar 2022
Chtita S, Ghamali M, Ousaa A, Aouidate A, Belhassan A, Taourati AI, Masand VH, Bouachrine M, Lakhlifi T (2019) QSAR study of anti-Human African trypanosomiasis activity for 2-phenylimidazopyridines derivatives using DFT and Lipinski’s descriptors. Heliyon 5(3):e01304
Chtita S, Aouidate A, Belhassan A, Ousaa A, Taourati AI, Elidrissi B, Ghamali M, Bouachrine M, Lakhlifi T (2020) QSAR study of N-substituted oseltamivir derivatives as potent avian influenza virus H5N1 inhibitors using quantum chemical descriptors and statistical methods. New J Chem 44:1747–1760
Elidrissi B, Ousaa A, Ghamali M, Chtita S, Ajana MA, Bouachrine M, Lakhlifi T (2014) Toxicity in vivo of nitro-aromatic compounds: DFT and QSAR results. J Comput Aided Mol Des 4:28–37
Daoui O, Elkhattabi S, Chtita S, Elkhalabi R, Zgou H, Benjelloun AT (2021) QSAR, Molecular Docking and ADMET properties in silico studies of Novel 4, 5, 6, 7-Tetrahydrobenzo [D]-Thiazol-2-Yl Derivatives Derived from Dimedone as potent Anti-Tumor agents through inhibition of C-Met receptor Tyrosine Kinase. Heliyon 7(7):E07463. https://doi.org/10.1016/j.heliyon.2021.e07463
Ouassaf M, Belaidi S, Benbrahim I, Belaidi H, Chtita S (2020) Quantitative structure-activity relationships of 1.2. 3 triazole derivatives as aromatase inhibition activity, Turkish Computational and Theoretical Chemistry 4(1):1–11. https://doi.org/10.33435/tcandtc.545369
Chtita S, Ghamali M, Hmamouchi R, Elidrissi B, Bourass M, Larif M, Bouachrine M, Lakhlifi T (2016) Investigation of antileishmanial activities of acridines derivatives against promastigotes and amastigotes form of parasites using quantitative structure activity relationship analysis, Advances in Physical Chemistry e5137289. https://doi.org/10.1155/2016/5137289
Diana G, Tommasi C (2002) Cross-validation methods in principal component analysis: a comparison. Stat Methods Appl 11:71–82
Chatterjee S, Hadi AS, Regression analysis by example, John Wiley & Sons, 2013
Rücker C, Rücker G, Meringer M (2007) Y-randomization–a useful tool in QSAR validation, or folklore. J Chem Inf Model 47:2345–2357
Eriksson L, Jaworska J, Worth AP, Cronin MT, McDowell RM, Gramatica P (2003) Methods for reliability and uncertainty assessment and for applicability evaluations of classification-and regression-based QSARs. Environ Health Perspect 111:1361–1375
Netzeva TI, Worth AP, Aldenberg T, Benigni R, Cronin MT, Gramatica P, Jaworska JS, Kahn S, Klopman G, Marchant CA (2005) Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships: the report and recommendations of ECVAM workshop 52. Altern Lab Anim 33:155–173
Sabando MV, Ponzoni I, Soto AJ (2019) Neural-based approaches to overcome feature selection and applicability domain in drug-related property prediction. Appl Soft Comput 85:105777
Jaworska J, Nikolova-Jeliazkova N, Aldenberg T (2005) QSAR applicability domain estimation by projection of the training set in descriptor space: a review. Altern Lab Anim 33:445–459
Zoete V, Daina A, Bovigny C, Michielin O (2016) SwissSimilarity: a web tool for low to ultra high throughput ligand-based virtual screening
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:1–13
U.N.E.C. for E. Secretariat, United Nations. Economic Commission for Europe. Secretariat. Globally Harmonized System of Classification and Labelling of Chemicals (GHS)., United Nations Publications, 2013.
Daoui O, Mazoir N, Bakhouch M, et al (2022) 3D-QSAR, ADME-Tox, and molecular docking of semisynthetic triterpene derivatives as antibacterial and insecticide agents. Struct Chem. https://doi.org/10.1007/s11224-022-01912-4
Shou WZ (2020) Current status and future directions of high-throughput ADME screening in drug discovery. Journal of Pharmaceutical Analysis 10:201–208
Chtita S, Belhassan A, Aouidate A, Belaidi S, Bouachrine M, Lakhlifi T (2021) Discovery of potent SARS-CoV-2 inhibitors from approved antiviral drugs via docking and virtual screening. Comb Chem High Throughput Screening 24:441–454
Ouassaf M, Belaidi S, Mogren Al Morgen M, Chtita S, Ullah Khan S, ThetHtar T (2021) Combined docking methods and molecular dynamics to identify effective antiviral 2, 5-diaminobenzophenonederivatives against SARS-CoV-2. J King Saud Univ Sci 33:101352. https://doi.org/10.1016/j.jksus.2021.101352
Brayer GD, Luo Y, Withers SG (1995) The structure of human pancreatic α-amylase at 1.8 Å resolution and comparisons with related enzymes. Protein Sci 4:1730–1742
Sybyl-X 2.0, Tripos International, St. Louis, Missouri, 63144, USA., Software Informer. (n.d.). https://sybyl-x.software.informer.com/2.0/. Accessed 26 Mar 2022
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461
Dassault Systèmes BIOVIA Discovery Studio Modeling Environment. Release 2017 Dassault Systèmes 2016., (n.d.). https://discover.3ds.com/discovery-studio-visualizer-download . Accessed 26 Mar 2022
H.J.C. Berendsen, B. Hess, E. Lindahl, D. Van Der Spoel, A.E. Mark, G. Groenhof, van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. (2005) GROMACS: fast, flexible, and free. J of Comput Chem 26(16):1701–1718. https://doi.org/10.1002/jcc.20291
Liao S-Y, Mo G-Q, Chen J-C, Zheng K-C (2014) Exploration of the binding mode between (-)-zampanolide and tubulin using docking and molecular dynamics simulation. J Mol Model 20:1–9
Sousa da Silva AW, Vranken WF (2012) ACPYPE-Antechamber python parser interface. BMC Res Notes 5:1–8
Brand W, Oosterhuis B, Krajcsi P, Barron D, Dionisi F, Van Bladeren PJ, Rietjens IM, Williamson G (2011) Interaction of hesperetin glucuronide conjugates with human BCRP, MRP2 and MRP3 as detected in membrane vesicles of overexpressing baculovirus-infected Sf9 cells. Biopharm Drug Dispos 32:530–535
Parrinello M, Rahman A, (2002) Polymorphic transítions in single crystals: a new molecular dynamics method polymorphic transítions in single crystals: a new molecular dynamics method
Kumari R., Kumar C, (2014) Open source drug discovery and A Lynn J Chem Inf Model 54:10–1021
Dahmani R, Manachou M, Belaidi S, Chtita S, Boughdiri S (2021) Structural characterization and QSAR modeling of 1,2,4-triazole derivatives as α-glucosidase inhibitors. New J Chem 45:1253–1261. https://doi.org/10.1039/D0NJ05298A
Chtita S, Belhassan A, Bakhouch M, Taourati AI, Aouidate A, Belaidi S, Moutaabbid M, Belaaouad S, Bouachrine M, Lakhlifi T (2021) QSAR study of unsymmetrical aromatic disulfides as potent avian SARS-CoV main protease inhibitors using quantum chemical descriptors and statistical methods. Chemom Intell Lab Syst 210:104266
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23:3–25
Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD (2002) Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 45:2615–2623
Ghose AK, Viswanadhan VN, Wendoloski JJ (1999) A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem 1:55–68
Szakács G, Váradi A, Özvegy-Laczka C, Sarkadi B (2008) The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME–Tox). Drug Discovery Today 13:379–393
Hollenberg PF (2002) Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev 34:17–35
Qais FA, Sarwar T, Ahmad I, Khan RA, Shahzad SA, Husain FM (2021) Glyburide inhibits non-enzymatic glycation of HSA: an approach for the management of AGEs associated diabetic complications. Int J Biol Macromol 169:143–152
Fouedjou RT, Chtita S, Bakhouch M, Belaidi S, Ouassaf M, Djoumbissie LA, Tapondjou LA, Qais FA (2021) Cameroonian medicinal plants as potential candidates of SARS-CoV-2 inhibitors. J Biomol Struct Dyn 0:1–15. https://doi.org/10.1080/07391102.2021.1914170
Rath B, Qais FA, Patro R, Mohapatra S, Sharma T (2021) Design, synthesis and molecular modeling studies of novel mesalamine linked coumarin for treatment of inflammatory bowel disease. Bioorganic Med Chem Lett 41:128029
Siddiqui S, Ameen F, Jahan I, Nayeem SM, Tabish M (2019) A comprehensive spectroscopic and computational investigation on the binding of the anti-asthmatic drug triamcinolone with serum albumin. New J Chem 43:4137–4151
Siddiqui S, Ameen F, Kausar T, Nayeem SM, Rehman SU, Tabish M (2021) Biophysical insight into the binding mechanism of doxofylline to bovine serum albumin: an in vitro and in silico approach. Spectrochim Acta Part A: Mol Biomol Spectrosc 249:119296
Author information
Authors and Affiliations
Contributions
Oussama Abchir: conceived and designed the experiments; performed the experiments; contributed reagents, materials, analysis tools, or data; and wrote the paper. Mebarka Ouassaf, and Faizan Abul Qais: conceived and designed the experiments; analyzed and interpreted the data; contributed reagents, materials, analysis tools, or data; and wrote the paper. Salah Belaidi and Said Belaaouad: conceived and designed the experiments. Ossama Daoui and Souad Elkhattabi: analyzed and interpreted the data and revised the paper. Samir Chtita: conceived and designed the experiments; analyzed and intereffects of diabetes on your bodpreted the data; contributed reagents, materials, analysis tools, or data; wrote the paper and supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Abchir, O., Daoui, O., Belaidi, S. et al. Design of novel benzimidazole derivatives as potential α-amylase inhibitors using QSAR, pharmacokinetics, molecular docking, and molecular dynamics simulation studies. J Mol Model 28, 106 (2022). https://doi.org/10.1007/s00894-022-05097-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05097-9