
Abstract
In this study, dimensional, conformational and dynamic behaviors of a short-chain branched styrene/1-octene copolymer chain with different 1-octene percentages, i.e., 0, 2, 4 and 6%, in toluene are investigated at the temperature of 298.15 K via molecular dynamics simulation. The chain dimensions and flexibility in the solvent are evaluated by calculating the radius of gyration (Rg), end-to-end distance (<r0>), surface area (Ach), and volume (Vch) of the copolymer chain. The mean square displacement (MSD) and diffusivity coefficient for each copolymer chain are measured to determine its dynamic behavior and mobility in aromatic media. To consider the effect of increasing the 1-octene co-monomer percentage on the copolymer chain affinity to the solvent molecules, the interaction energy (Eint) and Flory-Huggins (FH) interaction parameter are calculated for each equilibrated solution model. The simulation results indicate that the co-monomer level increment in the copolymer structure reduces the chain Rg amount and its interaction with the solvent. The <r0> of the chain increases up to 4% co-monomer content, while further co-monomer content decreases the <r0> value. Additionally, the viscosity of the equilibrated dilute solutions is calculated via non-equilibrium molecular dynamics simulation (NEMD). Moreover, the steric hindrance of the copolymers and the solvent molecules capturing in the dilute solution is determined via radial distribution function (RDF) analysis. Helmholtz free energy and the system entropy changes are calculated to evaluate the tendency of the copolymer to the solvent molecules and its dilute solution irregularity, respectively.

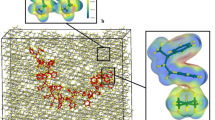
The figure shows the variations trend of the poly(styrene-co-1-octene) chain dimensions in toluene aromatic solvent by increasing the 1-octene content (x), after the equilibration state. Red and blue colors represent the carbon atoms of the copolymer chain backbone and 1-octene side chains, respectively. The styrene rings and the hydrogen atoms of the chains were removed for better view.














Similar content being viewed by others

References
Feldman D, Barbalata A (1996) Synthetic polymers: technology, properties, applications1st edn. Springer Science & Business Media, London
Mohammad F (2007) Specialty polymers: materials and applications. I. K. International Pvt Ltd, New Delhi
Nam C, Li H, Zhang G, Mike Chung TC (2016) Petrogel: new hydrocarbon (oil) absorbent based on polyolefin polymers. Macromolecules 49:5427–5437
Ceylan D, Dogu S, Karacik B, Yakan SD, Okay OS, Okay O (2009) Evaluation of butyl rubber as sorbent material for the removal of oil and polycyclic aromatic hydrocarbons from seawater. Environ Sci Technol 43:3846–3852
Tanobe VOA, Sydenstricker THD, Amico SC, Vargas JVC, Zawadzki SF (2008) Evaluation of flexible postconsumed polyurethane foams modified by polystyrene grafting as sorbent material for oil spills. J Appl Polym Sci 111:1842–1849
Lin J, Shang Y, Ding B, Yang J, Yu J, Al-Deyab SS (2012) Nanoporous polystyrene fibers for oil spill cleanup. Mar Pollut Bull 64:347–352
Zhou MH, Cho WJ (2002) Synthesis and properties of high oil-absorbent 4-tert- butylstyrene-EPDM divinylbenzene graft terpolymer. J Appl Polym Sci 85:2119–2129
Essawy HA, Essa MM, Abdeen Z (2010) Oil-absorptive polymeric networks based on dispersed oleophilized nanolayers of laponite within ethylene-propylene-diene monomer vulcanizates. J Appl Polym Sci 115:385–392
Shan GR, Xu PY, Weng ZX, Huang ZM (2003) Oil-absorption function of physical crosslinking in the high-oil-absorption resins. J Appl Polym Sci 90:3945–3950
Fouchet B (2009) Diffusion of mineral oil in styrene-butadiene polymer films. J Appl Polym Sci 111:2886–2891
Yuan X, Chung TCM (2012) Novel solution to oil spill recovery: using thermodegradable polyolefin oil superabsorbent polymer (Oil−SAP). Energy Fuel 26:4896–4902
Han J, Boyd RH (1996) Molecular packing and small-penetrant diffusion in polystyrene: a molecular dynamics simulation study. Polymer 37:1797–1804
Amani M, Iranagh SA, Golzar K, Sadeghi GMM, Modarress H (2014) Study of nanostructure characterizations and gas separation properties of poly(urethane–urea)s membranes by molecular dynamics simulation. J Membr Sci 462:28–41
Mozaffari F, Eslami H, Moghadasi J (2010) Molecular dynamics simulation of diffusion and permeation of gases in polystyrene. Polymer 51:300–307
Chen Y, Liu QL, Zhu AM, Zhang QG, Wu JY (2010) Molecular simulation of Co2/Ch4 permeabilities in polyamide–imide isomers. J Membr Sci 348:204–212
Nicholson LK, Higgins JS (1981) Dynamics of dilute polymer solutions. Macromolecules 14:836–843
Chen H, Li J, Ding Y, Zhang G, Zhang Q, Wu C (2005) Folding and unfolding of individual PNIPAM-g-PEO copolymer chains in dilute aqueous solutions. Macromolecules 38:4403–4408
Cheung DL (2012) Molecular simulation of hydrophobin adsorption at an oil−water interface. Langmuir 28:8730–8736
Ajori S, Ansari R, Darvizeh M (2016) On the vibrational behavior of single- and double-walled carbon nanotubes under the physical adsorption of biomolecules in the aqueous environment: a molecular dynamics study. J Mol Model 22:62–69
Haghighi S, Ansari R, Ajori S (2019) Interfacial properties of 3D metallic carbon nanostructures (T6 and T14)-reinforced polymer nanocomposites: a molecular dynamics study. J Mol Graph Model 92:341–356
Ansari R, Ajori S, Rouhi S (2014) Characterization of the mechanical properties of polyphenylene polymer using molecular dynamics simulations. Physica B 481:80–85
Haghighi S, Ansari R, Ajori S (2019) Influence of polyethylene cross-linked functionalization on the interfacial properties of carbon nanotube-reinforced polymer nanocomposites: a molecular dynamics study. J Mol Model 25:105–117
Ansari R, Rouhi S, Ajori S (2018) Molecular dynamics simulations of the polymer/amine functionalized single-walled carbon nanotubes interactions. Appl Surf Sci. https://doi.org/10.1016/j.apsusc.2018.04.133
Ansari R, Ajori S, Rouhi S (2015) Investigation of the adsorption of polymer chains on amine-functionalized double-walled carbon nanotubes. J Mol Model 21:312–322
Ajori S, Parsapour H, Ansari R (2019) Structural properties and buckling behavior of non-covalently functionalized single- and double-walled carbon nanotubes with pyrenelinked polyamide in aqueous environment using molecular dynamics simulations. J Phys Chem Solids 131:79–85
Tesei G, Paradossi G, Chiessi E (2012) Poly(vinyl alcohol) oligomer in dilute aqueous solution: a comparative molecular dynamics simulation study. J Phys Chem B 116:10008–10019
Horinaka J, Ito S, Yamamoto M, Matsuda T (2000) Molecular dynamics simulation of local motion of polystyrene chain end-comparison with the fluorescence depolarization study. Comput Theor Polym Sci 10:365–370
Dünweg B, Kremer K (1993) Molecular dynamics simulation of a polymer chain in solution. J Chem Phys 99:6983–6997
Drew PM, Adolf DB (2005) Intrinsic viscosity of dendrimers via equilibrium molecular dynamics. Soft Matter 1:146–151
Goicochea AG, Briseño M (2012) Application of molecular dynamics computer simulations to evaluate polymer-solvent interactions. J Coat Technol Res 9:279–286
Moe NE, Ediger MD (1995) Molecular dynamics computer simulation of polyisoprene local dynamics in dilute toluene solution. Macromolecules 28:2329–2338
Zhang B, Liu R, Zhang J, Liu B, He J (2016) MesoDyn simulation study of phase behavior for dye-polyether derivatives in aqueous solutions. Comput Theor Chem 1091:8–17
Zhelavskyi OS, Kyrychenko A (2019) Atomistic molecular dynamics simulations of the LCST conformational transition in poly(N-Vinylcaprolactam) in water. J Mol Graph Model. https://doi.org/10.1016/j.jmgm.2019.04.004
Mondello M, Yang HJ, Furuya H, Roe RJ (1994) Molecular dynamics simulation of atactic polystyrene. 1. Comparison with X-ray scattering data. Macromolecules 27:3566–3574
Tamaia Y, Fukuda M (2005) Sorption of organic solvents on the surface of crystalline syndiotactic polystyrene studied by molecular dynamics simulation. J Mol Struct 739:27–32
Oh SY, Bae YC (2012) Role of intermolecular interactions for upper and lower critical solution temperature behaviors in polymer solutions: molecular simulations and thermodynamic modeling. Polymer 53:3772–3779
Sun H (1994) Force field for computation of conformational energies, structures, and vibrational frequencies of aromatic polyesters. J Comput Chem 15:752–768
Sun H, Mumby SJ, Maple JR, Hagler AT (1994) An ab initio CFF93 all-atom force field for polycarbonates. J Am Chem Soc 116:2978–2987
Rasouli S, Moghbeli MR, Javan Nikkhah S (2018) A comprehensive molecular dynamics study of a single polystyrene chain in a good solvent. Curr Appl Phys 18:68–78
Plimpton S (1995) Fast parallel algorithms for short-range molecular dynamics. J Comput Phys 117:1–19
Gedde UW (1995) Polymer physics1st edn. Chapman and Hall, London
Lee SH (2011) Molecular dynamics simulation studies of viscosity and diffusion of n-alkane oligomers at high temperatures. Bull Kor Chem Soc 32:3909–3913
Han KH, Jeon GS, Hong IK, Lee SB (2013) Prediction of solubility parameter from intrinsic viscosity. J Ind Eng Chem 19:1130–1136
Frenkel D, Smit B (2001) Understanding molecular simulation: from algorithms to applications2nd edn. Academic Press, London
Martins SA, Sousa SF, Ramos MJ, Fernandes PA (2014) Prediction of solvation free energies with thermodynamic integration using the general amber force field. J Chem Theory Comput 10:3570–3577
McQuarrie DA (2000) Statistical mechanics. University Science Books, Saucilito
Mitchell MJ, McCammon JA (1991) Free energy difference calculations by thermodynamic integration: difficulties in obtaining a precise value. J Comput Chem 12:271–275
Resat H, Mezei M (1993) Studies on free energy calculations. I. Thermodynamic integration using a polynomial path. J Chem Phys 99:6052–6061
Gonçalves PFB, Stassen H (2003) Free energy of solvation from molecular dynamics simulations for low dielectric solvents. J Comput Chem 24:1758–1765
Gonçalves PFB, Stassen H (2004) Calculation of the free energy of solvation from molecular dynamics simulations. Pure Appl Chem 76:231–240
Zhang L, Wang X, Ma H, Huang Y (1999) Conformational behavior of short adsorbed polymer chains. Eur Polym J 35:167–172
Hansen CM (2000) Hansen solubility parameters: a User's handbook2nd edn. CRC Press, Boca Raton
Abe F, Einaga Y, Yoshizaki T, Yamakawa H (1993) Excluded-volume effects on the mean-square radius of gyration of oligo- and polystyrenes in dilute solutions. Macromolecules 26:1884–1890
Abe F, Einaga Y, Yamakawa H (1993) Excluded-volume effects on the intrinsic viscosity of oligomers and polymers of styrene and isobutylene. Macromolecules 26:1891–1897
Sperling LH (2006) Introduction to physical polymer science4th edn. Wiley, Hoboken
Yankova TS, Bobrovsky A, Vorobiev AK (2012) Order parameters <P2>, <P4> and <P6> of aligned nematic LC-polymer as determined by numerical simulation of EPR spectra. J Phys Chem B 116:6010–6016
Wheeler DR, Fuller NG, Rowley RL (1997) Non-equilibrium molecular dynamics simulation of the shear viscosity of liquid methanol: adaptation of the Ewald sum to lees± Edwards boundary conditions. Mol Phys 92:55–62
Wang BY, Cummings PT (1993) Non-equilibrium molecular dynamics calculation of the shear viscosity of carbon dioxide/Ethan mixtures. Mol Simul 10:1–11
Rasouli S, Moghbeli MR, Javan Nikkhah S (2019) A deep insight into the polystyrene chain in cyclohexane at theta temperature: molecular dynamics simulation and quantum chemical calculations. J Mol Model. https://doi.org/10.1007/s00894-019-4078-4
Prausnitz JM, Lichtenthaler RN, Azevedo EGD (1999) Molecular thermodynamics of fluid-phase equilibria3th edn. Prentice Hall PTR, Upper Saddle River
Bozdogan AE (2004) A method for determination of thermodynamic and solubility parameters of polymers from temperature and molecular weight dependence of intrinsic viscosity. Polymer 45:6415–6424
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rasouli, S., Moghbeli, M.R. & Nikkhah, S.J. Molecular dynamics simulation of polystyrene copolymer with octyl short-chain branches in toluene. J Mol Model 26, 80 (2020). https://doi.org/10.1007/s00894-020-4339-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-4339-2