
Abstract
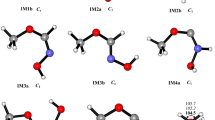
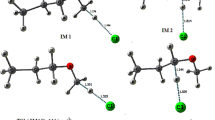
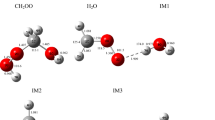
Mechanism of the reaction of CH3OCH2 with NO2 is explored theoretically at the M062X/MG3S and G4 levels. The calculated results indicate two stable association intermediates, CH3OCH2NO2 (IM1) and CH3OCH2ONO (IM2), which can be produced by the attack of the nitrogen or oxygen atom of NO2 to terminal carbon atom of CH3OCH2 without barrier involved. IM2 is found to take trans (IM2a)–cis (IM2b) conversion and isomerization to IM1, with the following stability order IM2a > IM2b > IM1. Starting from IM2a, the most feasible pathway is the direct O−NO bond cleavage leading to P1 (CH3OCH2O + NO) or the H-shift and O−NO bond rupture to produce P2 (CH3OCHO + HNO), both of which have comparable contribution to the title reaction. There also involves an H-transfer from the methyl group of IM2a to the N atom with the simultaneous dissociations of the C–O and O–N bonds to produce P4 (2CH2O + HNO). In addition, another dissociation pathway is open to IM2b which decompose to P5 (CH2O + CH3ONO) by the O−N and C−O bond scissions and the recombination of CH3O and NO. Because all the intermediates and transition states involved in the above pathways lie below reactants, the CH3OCH2 + NO2 reaction is expected to be rapid. Subsequent dissociation of IM1 and direct H-abstraction between CH3OCH2 and NO2 are kinetically almost inhibited due to significantly high barriers. The present results can lead us to deeply understand the mechanism of the title reaction and may be helpful for understanding NO2 combustion chemistry.





Similar content being viewed by others
Data availability
All data are included in this article and its supplementary material file.
References
Kohse-Höinghaus K, Oßwald P, Cool TA, Kasper T, Hansen N, Qi F, Westbrook CK, Westmoreland PR (2010) Biofuel combustion chemistry: from ethanol to biodiesel. Angew Chem Int Ed 49:3572–3597
Arcoumanis C, Bae C, Crookes R, Kinoshita E (2008) The potential of di-methyl ether (DME) as an alternative fuel for compression-ignition engines: a review. Fuel 87:1014–1030
Curran HJ, Pitz WJ, Westbrook CK, Dagaut P, Boettner J-C, Cathonnet M (1998) A wide range modeling study of dimethyl ether oxidation. Int J Chem Kinet 30:229–241
Peres R, Linnert HV (2004) Internal energy dependence of the unimolecular dissociation channels of dimethyl ether. Quim Nova 27:42–45
Miller JA, Bowman CT (1989) Mechanism and modeling of nitrogen chemistry in combustion. Prog Energy Combust Sci 15:287–338
Miller JA, Pilling MJ, Troe J (2005) Unravelling combustion mechanisms through a quantitative understanding of elementary reactions. Proc Combust Inst 30:43–88
Rissanen MP, Arppe SL, Timonen RS (2013) Kinetics of several oxygenated carbon-centered free radical reactions with NO2. J Phys Chem A 117:3902–3908
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA et al (2009) Gaussian 09, revision A.02. Gaussian, Inc., Wallingford
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Account 120:215–241
Lynch BJ, Zhao Y, Truhlar DG (2003) Effectiveness of diffuse basis functions for calculating relative energies by density functional theory. J Phys Chem A 107:1384–1388
Zheng J, Zhao Y, Truhlar DG (2009) The DBH24/08 database and its use to assess electronic structure model chemistries for chemical reaction barrier heights. J Chem Theory Comput 5:808–821
Hratchian HP, Schlegel HB (2004) Accurate reaction paths using a Hessian based predictor-corrector integrator. J Chem Phys 120:9918–9924
Hratchian HP, Schlegel HB (2005) Using hessian updating to increase the efficiency of a Hessian based predictor-corrector reaction path following method. J Chem Theory Comput 1:61–69
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83:735–746
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926
Domingo LR (2014) A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4:32415–32428
Jasiński R, Jasińska E, Dresler E (2017) A DFT computational study of the molecular mechanism of [3 + 2] cycloaddition reactions between nitroethene and benzonitrile N-oxides. J Mol Model 23:13/1–13/9
Wiberg KB (1968) Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 24:1083–1096
Alecu IM, Zheng J, Zhao Y, Truhlar DG (2010) Computational thermochemistry: scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries. J Chem Theory Comput 6:2872–2887
Curtiss LA, Redfern PC, Raghavachari K (2007) Gaussian-4 theory using reduced order perturbation theory. J Chem Phys 127:124105/1–124105/8
Johnson III RD (2019) Computational chemistry comparison and benchmark database, version 19; National Institute of Standards and Technology. http://cccbdb.nist.gov/ (accessed 19 May 2020)
Ruscic B, Bross DH (2017) Active thermochemical tables (ATcT) values based on version 1.122p of the thermochemical network, available at ATcT.anl.gov (accessed 23 August 2020)
Gray P (1955) Bond dissociation energies in nitrites and nitro compounds and the reaction of free radicals with nitrogen dioxide. Trans Faraday Soc 51:1367–1375
Domingo LR, Sáez JA (2009) Understanding the mechanism of polar Diels–Alder reactions. Org. Biomol Chem 7:3576–3583
Domingo LR, Chamorro E, Pérez P (2008) Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J Organomet Chem 73:4615–4624
Hammond GS (1955) A correlation of reaction rates. J Am Chem Soc 77:334–338
Arteca GA, Mezey PG (1988) Validity of the Hammond postulate and constraints on general one-dimensional reaction barriers. J Comput Chem 9:728–744
Funding
This study was funded by the National Natural Science Foundation of China under Grant no. 21606178.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Theoretical calculations, data collection, and analysis were performed by Yulei Guan, Xiangrui Meng, Xia Wang, and Liu Ru. The first draft of the manuscript was written by Yulei Guan and Haixia Ma. Review and editing were performed by Jirong Song. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Code availability
The copyrighted Gaussian software is available to us.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 105 kb)
Rights and permissions
About this article

Cite this article
Guan, Y., Meng, X., Wang, X. et al. Theoretical mechanistic study on the reaction of the methoxymethyl radical with nitrogen dioxide. J Mol Model 27, 18 (2021). https://doi.org/10.1007/s00894-020-04657-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-04657-1