
Abstract
Aldose reductase 2 (ALR2), which catalyzes the reduction of glucose to sorbitol using NADP as a cofactor, has been implicated in the etiology of secondary complications of diabetes. A pharmacophore model, Hypo1, was built based on 26 compounds with known ALR2-inhibiting activity values. Hypo1 contains important chemical features required for an ALR2 inhibitor, and demonstrates good predictive ability by having a high correlation coefficient (0.95) as well as the highest cost difference (128.44) and the lowest RMS deviation (1.02) among the ten pharmacophore models examined. Hypo1 was further validated by Fisher’s randomization method (95%), test set (r = 0.91), and the decoy set shows the goodness of fit (0.70). Furthermore, during virtual screening, Hypo1 was used as a 3D query to screen the NCI database, and the hit leads were sorted by applying Lipinski’s rule of five and ADME properties. The best-fitting leads were subjected to docking to identify a suitable orientation at the ALR2 active site. The molecule that showed the strongest interactions with the critical amino acids was used in molecular dynamics simulations to calculate its binding affinity to the candidate molecules. Thus, Hypo1 describes the key structure–activity relationship along with the estimated activities of ALR2 inhibitors. The hit molecules were searched against PubChem to find similar molecules with new scaffolds. Finally, four molecules were found to satisfy all of the chemical features and the geometric constraints of Hypo1, as well as to show good dock scores, PLPs and PMFs. Thus, we believe that Hypo1 facilitates the selection of novel scaffolds for ALR2, allowing new classes of ALR2 inhibitors to be designed.

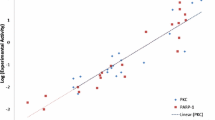
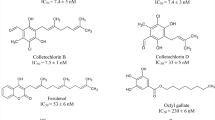
Pharmacophore model was generated and validated using various potent techniques. The validated Hypo1 was used in virtual screening to find a novel compounds and subjected to molecular docking and dynamics simulations











Similar content being viewed by others

Abbreviations
- ADME:
-
Absorption, distribution, metabolism, and excretion
- ALR2:
-
Aldose reductase 2
- BBB:
-
Blood–brain barrier
- DS:
-
Discovery Studio v.2.5
- EF:
-
Enrichment factor
- GF:
-
Goodness of fit
- HBA:
-
Hydrogen bond acceptor
- HAli:
-
Hydrophobic aliphatic
- HAro:
-
Hydrophobic aromatic
- MD:
-
Molecular dynamics
- NI:
-
Negative ionization
- NADPH:
-
Nicotinamide dinucleotide
- 53N:
-
3-[5-(3-Nitrophenyl) thiophen-2-yl] propanoic acid
- PME:
-
Particle mesh Ewald
- PLP:
-
Piecewise linear potential
- PMF:
-
Potential of mean force
- RA:
-
Ring aromatic
- RMS:
-
Root mean square
- RMSD:
-
Root mean square deviation
- RMSF:
-
Root mean square fluctuation
- TIM:
-
Triose phosphate isomerase
- VMD:
-
Visual molecular dynamics
References
National Institute of Diabetes and Digestive and Kidney Diseases (2011) National Diabetes Information Clearinghouse (NDIC). http://diabetes.niddk.nih.gov
Sato S, Kador PF (1990) Inhibition of aldehyde reductase by aldose reductase inhibitors. Biochem Pharmacol 40:1033–1042
Carper DA, Wistow G, Nishimura C, Graham C, Watanabe K, Fujii Y, Hayashi H, Hayaishi O (1989) A superfamily of NADPH-dependent reductases in eukaryotes and prokaryotes. Exp Eye Res 49:377–388
Costantino L, Rastelli G, Cignarella G, Vianello P, Barlocco D (1997) New aldose reductase inhibitors as potential agents for the prevention of long-term diabetic complications. Expert Opin Ther Patents 7:843–858
Singh SB, Malamas MS, Hohman TC, Nilakantan R, Carper DA, Kitchen D (2000) Molecular modeling of the aldose reductase–inhibitor complex based on the X-ray crystal structure and studies with single-site-directed mutants. J Med Chem 43:1062–1070
Nishimura C, Matsuura Y, Kokai Y, Akera T, Carper D, Morjana N, Lyons C, Flynn TG (1990) Cloning and expression of human aldose reductase. J Biol Chem 17:9788–9792
Sotriffer CA, Krämer O, Klebe G (2004) Probing flexibility and “induced-fit” phenomena in aldose reductase by comparative crystal structure analysis and molecular dynamics simulations. Proteins Struct Funct Bioinf 56:52–66
Fournier B, Bendeif EE, Bt G, Podjarny A, Lecomte C, Jelsch C (2009) Charge density and electrostatic interactions of fidarestat, an inhibitor of human aldose reductase. J Am Chem Soc 131:10929–10941. doi:10.1021/ja8095015
Gual P, Le Marchand-Brustel Y, Tanti JF (2003) Positive and negative regulation of glucose uptake by hyperosmotic stress. Diabetes Metab 29:566–575
Oates PJ (2002) Polyol pathway and diabetic peripheral neuropathy. Int Rev Neurobiol 50:325–328
Eisenmann M, Steuber H, Zentgraf M, Altenkamper M, Ortmann R, Perruchon J, Klebe G, Schlitzer M (2009) Structure-based optimization of aldose reductase inhibitors originating from virtual screening. Chem Med Chem 4:809–819
Mc VZ, Doan B, Dr S, Sredy J, Podjarny AD (2009) Discovery of [3-(4,5,7-trifluoro-benzothiazol-2-ylmethyl)-pyrrolo[2,3-b]pyridin-1-yl]acetic acids as highly potent and selective inhibitors of aldose reductase for treatment of chronic diabetic complications. Bioorg Med Chem Lett 7:2006–2008
Petrova T, Lunin VY, Ginell S, Hazemann I, Lazarski K, Mitschler A, Podjarny A, Joachimiak A (2009) X-ray-radiation-induced cooperative atomic movements in protein. J Mol Biol 387:1092–1105
El-Kabbani O, Darmanin C, Schneider TR, Hazemann I, Ruiz F, Oka M, Joachimiak A, Schulze-Briese C, Tomizaki T, Mitschler A, Podjarny A (2004) Ultrahigh resolution drug design II. Atomic resolution structures of human aldose reductase holoenzyme complexed with fidarestat and minalrestat: implications for the binding of cyclic imide inhibitors. Proteins Struct Funct Bioinf 55:805–813
El-Kabbani O, Dk W, Petrash M, Quiocho FA (1998) Structural features of the aldose reductase and aldehyde reductase inhibitor-binding sites. Mol Vision 4:19–25
Oka M, Matsumoto Y, Sugiyama S, Tsuruta N, Matsushima M (2000) A potent aldose reductase inhibitor, (2S,4S)-6-fluoro-2′,5′-dioxospiro[chroman-4,4′-imidazolidine]-2-carboxamide (fidarestat): its absolute configuration and interactions with the aldose reductase by X-ray crystallography. J Med Chem 43:2479–2483
Steuber H, Zentgraf M, Podjarny A, Heine A, Klebe G (2006) High-resolution crystal structure of aldose reductase complexed with the novel sulfonyl-pyridazinone inhibitor exhibiting an alternative active site anchoring group. J Mol Biol 356:45–56
Ramana KV, Srivastava SK (2010) Aldose reductase: a novel therapeutic target for inflammatory pathologies. Int J Biochem Cell Biol 42:17–20
Miyamoto S (2002) Molecular modeling and structure-based drug discovery studies of aldose reductase inhibitors. Chem-Bio Inf J 2:74–85
Accelrys Software, Inc. (2011) Corporate homepage. http://www.accelrys.com
Mylari BL, Larson ER, Beyer TA, Zembrowski WJ, Aldinger CE, Dee MF, Siegel TW, Singleton DH (1991) Novel, potent aldose reductase inhibitors: 3,4-dihydro-4-oxo-3-[[5-(trifluoromethyl)-2-benzothiazolyl]methyl]-1-phthalazineacetic acid (zopolrestat) and congeners. J Med Chem 34:108–122
Mylari BL, Zembrowski WJ, Beyer TA, Aldinger CE, Siegel TW (1992) Orally active aldose reductase inhibitors: indazoleacetic, oxopyridazineacetic, and oxopyridopyridazineacetic acid derivatives. J Med Chem 35:2155–2162
Lipinski CA, Aldinger CE, Beyer TA, Bordner J, Burdi DF, Bussolotti DL, Inskeep PB, Siegel TW (1992) Hydantoin bioisosteres. In vivo active spiro hydroxy acetic acid aldose reductase inhibitors. J Med Chem 35:2169–2177
Advanced Chemistry Development, Inc. (2010) Homepage. http://www.acdlabs.com
Brooks BR, Brooks CL 3rd, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30:1545–1614
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4:187–217
MacKerell AD, Brooks CL, Nilsson L, Roux B, Won Y, Karplus M (1998) CHARMM: the energy function and its paramerterization with an overview of the program. In: Schleyer (ed) The encyclopedia of computational chemistry. Wiley, Chichester, pp 271–277
Smellie A, Teig SL, Towbin P (1995) Poling: promoting conformational variation. J Comput Chem 16:171–187
Sakkiah S, Thangapandian S, John S, Lee KW (2011) Pharmacophore based virtual screening, molecular docking studies to design potent heat shock protein 90 inhibitors. Eur J Med Chem 46:2937–2947
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26
Sakkiah S, Thangapandian S, John S, Kwon YJ, Lee KW (2010) 3D QSAR pharmacophore based virtual screening and molecular docking for identification of potential HSP90 inhibitors. Eur J Med Chem 45:2132–2140
Berman HM (2008) The Protein Data Bank: a historical perspective. Acta Crystallogr A 64:88–95
The PDB Team (2003) The Protein Data Bank. Methods Biochem Anal 44:181–198
Berendsen HJC, van der Spoel D, van Drunen R (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun 91:43–56
Spoel DVD, Patriksson A, Seibert MM (2007) Protein folding properties from molecular dynamics simulations. In: Proc 8th Int Conf on Applied Parallel Computing: State of the Art in Scientific Computing (PARA 2006), Umeå, Sweden, 18–21 June 2006
Schuttelkopf AW, van Aalten DMF (2004) PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr D 60:1355–1363
Aalten DMF, Bywater R, Findlay JBC, Hendlich M, Hooft RWW, Vriend G (1996) PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J Comput Aided Mol Des 10:255–262
Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N.log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Hess B, Bekker H, Berendsen H, Fraaije J (1997) LINCS: A linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472
Venkatachalam CM, Jiang X, Oldfield T, Waldman M (2003) LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graph Model 21:289–307
Acknowledgments
This research was supported by the Basic Science Research Program (2009–0073267), the Pioneer Research Center Program (2009–0081539), and the Management of Climate Change Program (2010–0029084) through the National Research Foundation of Korea (NRF), as funded by the Ministry of Education, Science and Technology (MEST) of the Republic of Korea. This work was also supported by the Next-Generation BioGreen21 Program (PJ008038) from the Rural Development Administration (RDA) of the Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sakkiah, S., Thangapandian, S. & Lee, K.W. Pharmacophore modeling, molecular docking, and molecular dynamics simulation approaches for identifying new lead compounds for inhibiting aldose reductase 2. J Mol Model 18, 3267–3282 (2012). https://doi.org/10.1007/s00894-011-1247-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-011-1247-5