
Abstract
A novel molecular shape similarity comparison method, namely SHeMS, derived from spherical harmonic (SH) expansion, is presented in this study. Through weight optimization using genetic algorithms for a customized reference set, the optimal combination of weights for the translationally and rotationally invariant (TRI) SH shape descriptor, which can specifically and effectively distinguish overall and detailed shape features according to the molecular surface, is obtained for each molecule. This method features two key aspects: firstly, the SH expansion coefficients from different bands are weighted to calculate similarity, leading to a distinct contribution of overall and detailed features to the final score, and thus can be better tailored for each specific system under consideration. Secondly, the reference set for optimization can be totally configured by the user, which produces great flexibility, allowing system-specific and customized comparisons. The directory of useful decoys (DUD) database was adopted to validate and test our method, and principal component analysis (PCA) reveals that SH descriptors for shape comparison preserve sufficient information to separate actives from decoys. The results of virtual screening indicate that the proposed method based on optimal SH descriptor weight combinations represents a great improvement in performance over original SH (OSH) and ultra-fast shape recognition (USR) methods, and is comparable to many other popular methods. Through combining efficient shape similarity comparison with SH expansion method, and other aspects such as chemical and pharmacophore features, SHeMS can play a significant role in this field and can be applied practically to virtual screening by means of similarity comparison with 3D shapes of known active compounds or the binding pockets of target proteins.

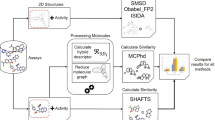
Schematic diagram of spherical harmonic (SH) based weighted similarity calculation. First, molecular surfaces are projected to groups of SH producing a series of projection coefficients which are used to calculate SH descriptors. Then, a genetic algorithm based searching process will be carried out, producing a group of optimal weights which can best separate actives from negatives. Finally, combining the SH descriptor and corresponding weights, a similarity score was calculated and used to rank the candidate molecules








Similar content being viewed by others

References
Rester U (2008) From virtuality to reality - Virtual screening in lead discovery and lead optimization: a medicinal chemistry perspective. Curr Opin Drug Discov Dev 11:559–568
Shoichet BK (2004) Virtual screening of chemical libraries. Nature 432:862–865
Kirchmair J, Markt P, Distinto S, Schuster D, Spitzer GM, Liedl KR, Langer T, Wolber G (2008) The Protein Data Bank (PDB), its related services and software tools as key components for in silico guided drug discovery. J Med Chem 51:7021–7040
Hristozov DP, Oprea TI, Gasteiger J (2007) Virtual screening applications: a study of ligand-based methods and different structure representations in four different scenarios. J Comput Aided Mol Des 21:617–640
Waszkowycz B (2008) Towards improving compound selection in structure-based virtual screening. Drug Discov Today 13:219–226
Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3:935–949
Lengauer T, Rarey M (1996) Computational methods for biomolecular docking. Curr Opin Struct Biol 6:402–406
Evers A, Hessler G, Matter H, Klabunde T (2005) Virtual screening of biogenic amine-binding G-protein coupled receptors: Comparative evaluation of protein- and ligand-based virtual screening protocols. J Med Chem 48:5448–5465
Hawkins PCD, Skillman AG, Nicholls A (2007) Comparison of shape-matching and docking as virtual screening tools. J Med Chem 50:74–82
McGaughey GB, Sheridan RP, Bayly CI, Culberson JC, Kreatsoulas C, Lindsley S, Maiorov V, Truchon JF, Cornell WD (2007) Comparison of topological, shape, and docking methods in virtual screening. J Chem Inf Model 47:1504–1519
Perez-Nueno VI, Ritchie DW, Rabal O, Pascual R, Borrell JI, Teixido J (2008) Comparison of ligand-based and receptor-based virtual screening of HIV entry inhibitors for the CXCR4 and CCR5 receptors using 3D ligand shape matching and ligand-receptor docking. J Chem Inf Model 48:509–533
Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS (2006) A critical assessment of docking programs and scoring functions. J Med Chem 49:5912–5931
Tirado-Rives J, Jorgensen WL (2006) Contribution of conformer focusing to the uncertainty in predicting free energies for protein-ligand binding. J Med Chem 49:5880–5884
Gohlke H, Klebe G (2002) Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew Chem Int Ed Engl 41:2645–2676
Daylight Chemical Information Systems, Inc. Daylight Chemical Information Systems Inc. http://www.daylight.com. Accessed 1 May 2010
Tripos, Inc. http://www.tripos.com. Accessed 1 May 2010
Durant JL, Leland BA, Henry DR, Nourse JG (2002) Reoptimization of MDL keys for use in drug discovery. J Chem Inf Comput Sci 42:1273–1280
Cheeseright TJ, Mackey MD, Melville JL, Vinter JG (2008) FieldScreen: Virtual Screening Using Molecular Fields. Application to the DUD Data Set. J Chem Inf Model 48:2108–2117
Cheeseright T, Mackey M, Rose S, Vinter A (2006) Molecular field extrema as descriptors of biological activity: definition and validation. J Chem Inf Model 46:665–676
Marin RM, Aguirre NF, Daza EE (2008) Graph theoretical similarity approach to compare molecular electrostatic potentials. J Chem Inf Model 48:109–118
Ronkko T, Tervo AJ, Parkkinen J, Poso A (2006) BRUTUS: optimization of a grid-based similarity function for rigid-body molecular superposition. II. Description and characterization. J Comput Aided Mol Des 20:227–236
Thorner DA, Willett P, Wright PM, Taylor R (1997) Similarity searching in files of three-dimensional chemical structures: representation and searching of molecular electrostatic potentials using field-graphs. J Comput Aided Mol Des 11:163–174
Vainio MJ, Puranen JS, Johnson MS (2009) ShaEP: molecular overlay based on shape and electrostatic potential. J Chem Inf Model 49:492–502
Good AC, Hodgkin EE, Richards WG (1992) The utilisation of Gaussian functions for the rapid evaluation of molecular similarity. J Comput Inf Comput Sci 32:188–191
Grant JA, Gallardo MA, Pickup BT (1996) A fast method of molecular shape comparison: a simple application of Gaussian descriptor of molecular shape. J Comput Chem 17:1653–1666
Ballester PJ, Richards WG (2007) Ultrafast shape recognition to search compound databases for similar molecular shapes. J Comput Chem 28:1711–1723
Ballester PJ, Finn PW, Richards WG (2009) Ultrafast shape recognition: evaluating a new ligand-based virtual screening technology. J Mol Graph Model 27:836–845
Ballester PJ (2011) Ultrafast shape recognition: method and applications. Future Med Chem 3:65–78
Mavridis L, Hudson BD, Ritchie DW (2007) Toward high throughput 3D virtual screening using spherical harmonic surface representations. J Chem Inf Model 47:1787–1796
Cai WS, Shao XG, Maigret B (2002) Protein-ligand recognition using spherical harmonic molecular surfaces: towards a fast and efficient filter for large virtual throughput screening. J Mol Graph Model 20:313–328
Morris RJ, Najmanovich RJ, Kahraman A, Thornton JM (2005) Real spherical harmonic expansion coefficients as 3D shape descriptors for protein binding pocket and ligand comparisons. Bioinformatics 21:2347–2355
Jakobi AJ, Mauser H, Clark T (2008) ParaFrag–an approach for surface-based similarity comparison of molecular fragments. J Mol Model 14:547–558
DiMaio FP, Soni AB, Phillips GN, Shavlik JW (2009) Spherical-harmonic decomposition for molecular recognition in electron-density maps. Int J Data Min Bioinform 3:205–227
Huang N, Shoichet BK, Irwin JJ (2006) Benchmarking sets for molecular docking. J Med Chem 49:6789–6801
A directory of useful decoys. http://dud.docking.org/. Accessed 13 June 2010
Jahn A, Hinselmann G, Fechner N, Zell A (2009) Optimal assignment methods for ligand-based virtual screening. J Cheminform 1:14
Good AC, Oprea TI (2008) Optimization of CAMD techniques 3. Virtual screening enrichment studies: a help or hindrance in tool selection? J Comput Aided Mol Des 22:169–178
Rogers D, Hahn M (2010) Extended-connectivity fingerprints. J Chem Inf Model 50:742–754
Accelrys, Inc. http://accelrys.com/. Accessed 1 May 2010
Ritchie DW, Kemp GJL (1999) Fast computation, rotation, and comparison of low resolution spherical harmonic molecular surfaces. J Comput Chem 20:383–395
Connolly ML (1983) Solvent-accessible surfaces of proteins and nucleic acids. Science 221(4612):709–713
Laurikkala J (2001) Improving identification of difficult small classes by balancing class distribution. Lecture Notes Comput Sci 2101:63–66
Lee Y, Jeon K, Lee JT, Kim S, Kim VN (2002) MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 21:4663–4670
Nicholls A (2008) What do we know and when do we know it? J Comput Aided Mol Des 22:239–255
Truchon JF, Bayly CI (2007) Evaluating virtual screening methods: good and bad metrics for the "early recognition" problem. J Chem Inf Model 47:488–508
Nicholls A, McGaughey GB, Sheridan RP, Good AC, Warren G, Mathieu M, Muchmore SW, Brown SP, Grant JA, Haigh JA, Nevins N, Jain AN, Kelley B (2010) Molecular shape and medicinal chemistry: a perspective. J Med Chem 53:3862–3886
Kahraman A, Morris RJ, Laskowski RA, Thornton JM (2007) Shape variation in protein binding pockets and their ligands. J Mol Biol 368:283–301
Kirchmair J, Markt P, Distinto S, Wolber G, Langer T (2008) Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection–what can we learn from earlier mistakes? J Comput Aided Mol Des 22:213–228
Jain AN, Nicholls A (2008) Recommendations for evaluation of computational methods. J Comput Aided Mol Des 22:133–139
Irwin JJ, Shoichet BK (2005) ZINC—a free database of commercially available compounds for virtual screening. J Chem Inf Model 45:177–182
Acknowledgments
This work was supported by the Special Fund for Major State Basic Research Project (grant 2009CB918501), the National Natural Science Foundation of China (grants 20803022), the Shanghai Committee of Science and Technology (grants 09dZ1975700 and 10431902600), the 863 Hi-Tech Program of China (grant 2007AA02Z304), and the Major National Scientific and Technological Project of China (grant 2009ZX09501-001). H.L. is also sponsored by Shanghai Rising-Star Program (grant 10QA1401800) and the Fundamental Research Funds for the Central Universities. The program and test sets of SHeMS are available from H.L. upon request.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Author contributions
C.C. designed and validated the Cyndi method, and also contributed to analysis and data interpretation and co-drafted the manuscript with J.G. and X.L. J.G. contributed to the design of method. X.L. contributed to method validation. H.L. conceived the idea of the SHeMS and provided direction for its development and revised the subsequent drafts of this manuscript with D.G., and H.J. All authors read and approved the final manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material
Supplementary file 1 – ESM_1.pdf
This file contains the ZINC codes of molecules passed pre-process and used for weights optimization. (PDF 183 kb)
Rights and permissions
About this article
Cite this article
Cai, C., Gong, J., Liu, X. et al. A novel, customizable and optimizable parameter method using spherical harmonics for molecular shape similarity comparisons. J Mol Model 18, 1597–1610 (2012). https://doi.org/10.1007/s00894-011-1173-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-011-1173-6