
Abstract
Magnetic properties of five samples obtained in reactions between nFeVO4 and (1 − n)Co3V2O8 (with the composition index n = 0.54, 0.50, 0.40, 0.30 and 0.20) have been investigated by dc magnetometry and electron paramagnetic resonance (EPR). The investigated samples contained two phases: one having lyonsite-type structure and the other Co3V2O8. Temperature dependence of dc magnetic susceptibility showed the Curie–Weiss paramagnetic behaviour with a strong antiferromagnetic (AFM) interaction in the high-temperature range leading to magnetic transitions at 6 and 12 K. For all samples, the magnetic hysteresis loop in isothermal magnetisation was registered at T = 5 K indicating the existence of a ferromagnetic component. Samples n = 0.54 and 0.50 displayed a large coercive field and remanent magnetisation in contrast to the other three samples due to higher content of the lyonsite-type structure. EPR spectra of the studied samples showed a broad asymmetric line that was decomposed on lorentzian or gaussian components. Temperature dependence of the spectral parameters (resonance field, linewidth, integrated intensity) of these components was studied in high-temperature range (T > 90 K). The observed spectra were attributed to different types of magnetic spin clusters.
Similar content being viewed by others

1 Introduction
Phases of the lyonsite- or howardevansite-type (abbreviated to L-type and H-type, respectively) structures contain isolated VO4 tetrahedrons which build a lattice of orthovanadates (V) of divalent metals [1]. These in turn are connected with strong catalytic activity in the reaction of the oxidizing dehydrogenation of the saturated to unsaturated compounds [2]. From this point of view, the FeVO4–Co3V2O8 system is an interesting object of investigation because in samples from that system two phases of the L-type and the H-type structures have been detected [3,4,5]. The L-type structure can accommodate cationic mixing as well as cationic vacancies therefore a large number of chemical compounds can adopt this structure type. The H-type structure is found in the products of contemporary fumarole activity of few volcanoes [6].
The starting compounds, FeVO4 and Co3V2O8, have very interesting magnetic properties, especially in the low-temperature range. Triclinic iron vanadate FeVO4 large single crystals displayed two magnetic orderings at 20 and 13 K related to two different Fe ligand environments of octahedral FeO6 and trigonal bipyramidal FeO5 in a six-column doubly bent chain [7]. The nanocrystalline FeVO4 prepared by co-precipitation method has shown similar transitions from paramagnetic to antiferromagnetic (AFM) state at 14 and 19.9 K [8]. Magnetic interactions among three different Fe cation sites in FeVO4 are responsible for large magnetic frustration evidenced by a negative and large Curie–Weiss temperature TCW = − 125 K in paramagnetic phase [9]. Co3V2O8 compound is an example of material in which the geometrically frustrated buckled kagome lattice is realised [10,11,12,13,14,15,16]. The kagome staircase lattice of Co3V2O8 contains Co2+ (S = 3/2) ions and complicated interplay of magnetic interactions yields a rich magnetic phase diagram. The material is paramagnetic at high temperature (above 11.3 K), and ferromagnetic (FM) at low temperature (below 6 K) or under a large applied field. Between these temperatures different AFM spin density wave phases (incommensurate and commensurate) have been detected [12, 13].
Magnetic properties of a number of phases from the FeVO4–Co3V2O8 system have been already studied using dc magnetometry and EPR/FMR (electron paramagnetic resonance/ferromagnetic resonance) techniques [17,18,19,20]. In Ref. [17], the EPR spectra of twenty samples from FeVO4–Co3V2O8 system at room temperature (RT) were presented. The spectra displayed a strong dependence on initial concentrations of FeVO4 and Co3V2O8 and large differences in spectra intensity. In Refs. [18,19,20], more detailed magnetic investigations of samples from the nFeVO4/(1 − n)Co3V2O8 system (where n is the composition index, 0 < n < 1) were presented. In Ref. [18], magnetic study of samples with the composition indexes n = 0.81 and 0.83 was published, in Ref. [19] samples with n = 0.96, 0.86, 0.84 and 0.83 have been investigated, while in Ref. [20] samples with n = 0.82, 0.80, 0.78 and 0.76 were discussed. A very intricate nature of magnetism due to a mixture of phases, complicated crystal structure and competition of magnetic interactions was demonstrated. In the high temperature, a strong AFM interaction was registered which depended on the concentration of FeVO4 in initial mixture. At low temperature, the hysteresis loops were observed for these compounds and loop parameters were strongly dependent on initial mixture content.
The aim of the present study is to examine the magnetic properties of five samples from the FeVO4–Co3V2O8 system using dc magnetometry and EPR techniques. In the current samples, the content of FeVO4 in initial mixtures is smaller (and Co3V2O8 bigger) than in previously published papers. The present study is thus a further step towards a full magnetic characterisation of all phases synthesized in the FeVO4–Co3V2O8 system. Despite belonging to the same system they show a great diversity of their physical and chemical properties, therefore, a thorough investigation of all phases is needed for comprehensive understanding of mechanism responsible for that multifaceted behaviour. Due to a complicated crystal structure and disorder introduced by randomness in metallic sites occupation, the magnetic frustration is expected to appear in these compounds as a result of competition between localised spins on a lattice interacting through various exchange pathways that cannot be simultaneously satisfied. This is an intensively studied subject in magnetism and the presented in this paper EPR and magnetometric studies of few compounds from the FeVO4–Co3V2O8 system could provide a fertile ground for observation of these exciting phenomena.
2 Experimental
Five investigated samples were synthesized by the solid-state reaction method. The initial mixtures, containing cobalt(II) orthovanadate(V) (Co3V2O8) and iron(III) orthovanadate(V) (FeVO4), were homogenized and then were heated in air in four stages, i.e. 680 °C (20 h) + 780 °C (20 h) + 920 °C (20 h) + 920 °C (20 h). The source of the present samples is the same as before and their basic chemical characteristics are presented in Ref. [5]. In that paper, samples were numbered 1–24, therefore the labelling of the current samples will be continued. The samples in this paper will be designated as S19, S20, S21, S22, and S23 (they were numbered as 19, 20, 21, 22, and 23 in Ref. [5]). The composition index n of these five samples is n = 0.54, 0.50, 0.40, 0.30, 0.20, correspondingly. Composition of the initial mixtures is presented in Table 1. After each heating stage samples were gradually cooled down in the furnace to RT and, after grinding, their compositions were checked by XRD analysis. The powder diffraction patterns of samples were recorded with the Empyrean II diffractometer (PANalytical) with Cu radiation. The identification of phases present in the samples was conducted on the basis of its XRD characteristics contained in the PDF card 04-014-2048 (the L-type phase) and 04-014-1635 (Co3V2O8).
DC magnetization measurements were carried out using an MPMS-7 SQUID magnetometer in 2–300 K temperature range and in magnetic fields up to 70 kOe in the zero-field cooling (ZFC) and field cooling (FC) modes. Standard X-band Bruker E 500 spectrometer (ν = 9.4 GHz) with magnetic field modulation of 100 kHz was used to record EPR spectra. Temperature EPR measurements were performed in the 90–290 K range using an Oxford nitrogen-flow cryostat.
3 Results and Discussion
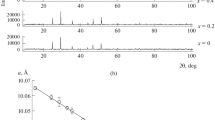
XRD results of the investigated samples, obtained after their final heating stage, showed that they contained two phases: the L-type phase and Co3V2O8 phase (see Table 1). Figure 1 displays L-type phase and Co3V2O8 structures. Figure 2 presents a part of XRD pattern of sample S21 as a representative of the investigated samples. The content of Co3V2O8 in the obtained mixtures increases from sample S19 to sample S23, whereas the content of L-type phase decreases from sample S19 to sample S23.

L-type phase (a) and Co3V2O8 (b) structures

XRD pattern of sample S21 with lines from L-type structure (designated by L) and Co3V2O8 (designated by *) phases
Figure 3 shows the temperature dependence of dc magnetic susceptibility χ(T) (here χ is defined as χ = M/H, where M is magnetisation and H an external magnetic field) and the reciprocal magnetic susceptibility χ−1(T) of the five studied samples registered in an external magnetic field H = 500 Oe in ZFC and FC modes. In high temperature range (T > 200 K), a linear dependence of χ−1(T) is observed. Consequently, χ(T) in that temperature range could be described by the Curie–Weiss law:
where Cmol is the molar Curie constant, and TCW is the Curie–Weiss temperature. From the molar Curie constant Cmol, the effective magnetic moment μeff (in units of Bohr magnetons) could be calculated:
where kB is the Boltzmann constant, NA is the Avogadro constant, and µB is the Bohr magneton. Because the studied samples contain different phases with unknown relative concentration, instead of usually used formula unit (f.u.), the known initial mixture unit (i.m.u.) will be used [20]. The mass of [i.m.u.] is calculated as
where z is the molar fraction (mol%) of FeVO4 in an initial mixture (0 < z < 1, see Table 1). The Curie constant can be recalculated, using instead of a gram, the mass of [i.m.u.]. The effective magnetic moment of [i.m.u.] calculated from Eq. (2) represents the magnetic moment of a compound calculated for z iron ions and 3(1 − z) cobalt ions. The results of calculations of TCW and the effective magnetic moment of [i.m.u.] in the high-temperature range are presented graphically in Fig. 4. For all five samples, a negative value of TCW is calculated. It indicates the presence of a strong AFM interaction between paramagnetic ions. As the strength of this AFM interaction is concerned it seems to diminish going from sample S19–S21, but increases on further Co content increase in sample S22 and S23.

Temperature dependence of the dc magnetic susceptibility χ (left axis) and the reciprocal magnetic susceptibility 1/χ (right axis) measured in an external magnetic field H = 500 Oe in ZFC and FC modes

Curie–Weiss temperature and the effective magnetic moment calculated from the Curie–Weiss law in high-temperature range
The obtained values of the effective magnetic moments (Fig. 4, horizontal axis) could be compared with theoretical values expected for a mixture of high-spin Fe and Co ions. The latter could be calculated from the following equation:
where μ1eff is an effective moment of Fe ion, and μ2eff is an effective moment of Co ion (in units of µB). For a single Fe3+ ion (S = 5/2) μ1eff = 5.9 μB, while for Co2+ ion (S = 3/2) μ2eff = 3.9 μB. Equation (4) provides the effective magnetic moment in the range from 6.29 to 6.56 µB for our samples, increasing with cobalt contents (see Fig. 5). As can be seen in Fig. 5, the experimentally determined values of µeff (points) are bigger than calculated from Eq. (4) (bars). The difference between experimental and theoretical values could be explained by bigger than assumed g-factors (due to additional orbital contribution) of both paramagnetic ions and by contribution of defect V4+ (S = ½) ions.

Experimental effective magnetic moments (points) and calculated effective magnetic moments (bars) assuming high-spin iron and cobalt ions
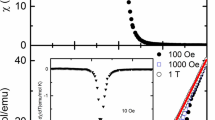
Figure 6 (upper panel) presents the temperature dependence of magnetic susceptibility in ZFC mode in the low temperature range, measured in an external magnetic field H = 10 Oe. To see the changes in χ(T) more clearly, the lower panel in Fig. 6 presents the temperature dependence of \(\frac{{{\text{d}}\chi_{\text{ZFC}} }}{{{\text{d}}T}}\) derivative. As the shape of χ(T) is concerned, in all studied sample it is similar with two maxima at two different temperatures: one at 12 K (very weak) and the other at 6 K (much stronger). The strength of these maxima increases with cobalt contents. As the observed two transition temperatures (6 and 12 K) are very close to the ones observed in Co3V2O8 compound (6 and 11.3 K), it seems that this phase is responsible for the observed magnetometric response.

Temperature dependence of the dc magnetic susceptibility χ in the low temperature range in ZFC mode in H = 10 Oe external magnetic field (upper panel) and the temperature gradient of the magnetic susceptibility dχ/dT (lower panel)
Figure 7 shows the temperature dependence of susceptibilities difference (χFC − χZFC) measured in an external magnetic field H = 10 Oe. The difference between magnetisation in FC and ZFC modes is mainly due to the existence of energy barriers of magnetic anisotropy [21]. All magnetic moments participate in magnetisation in FC mode, while in ZFC modes only these moments contribute to magnetisation of which the energy barriers are overcame by thermal energy at the measuring temperature. Examination of the susceptibilities differences in our samples revealed that the magnetic anisotropy is very similar with the exception of sample S20 in which it is much bigger. The susceptibility difference in ZFC and FC modes is zero above 12 K, it increases with temperature decrease down to 6 K and starts to increase significantly below that temperature. Both temperatures at which (χFC − χZFC) are subjected to characteristic changes coincide with the transition temperatures in Co3V2O8 phase.

Temperature dependence of susceptibility difference (χFC − χZFC) measured in an external magnetic field H = 10 Oe
Figure 8 presents isothermal magnetisation M(H) of five samples registered at 5 K. The dependence of magnetisation on external magnetic field is not linear as is expected for paramagnetic and AFM materials. M(H) curves show a visible curvature usually registered for FM materials. The right panel in Fig. 8 displays in an extended scale the central part of M(H) dependence, showing a part of the hysteresis loop. The occurrence of a hysteresis loop in isothermal magnetisation indicates there is a FM component in all investigated samples. The values of two parameters of a hysteresis loop: Hc—the coercive field, and Mr—the remanent magnetisation (remanence), for all samples measured at T = 5 K are presented in Fig. 9. Both Hc and Mr seem to be positively correlated—an increase of the coercive field is related to an increase of the remanence. Both parameters also decrease with the increase of Co3V2O8 content in an initial mixture what suggests an important role of Fe ions in development of hysteresis loops. Considering the obtained values of Hc and Mr all five investigated samples can be divided into two groups: those with small values of these parameters (samples S21, S22, and S23, with Hc < 100 Oe, Mr < 1.3 emu/g), and with larger ones (samples S19, S20, with Hc ~ 1000 Oe, Mr > 3 emu/g). In samples of the former group, a smaller part of magnetic ions seems to be involved in the FM component. This might be correlated with a higher content of Co3V2O8 phase evidenced by XRD method. Another possible explanation of the FM component is the presence of residual metallic Fe or Co impurities in our samples. Trace amounts of them would not be visible in XRD spectra but might influence catalytic activities of these samples.

Isothermal magnetisations at T = 5 K in a broad magnetic field range (left panel) and the central part of hysteresis loops in an extended scale (right panel)

Coercive field and the remanent magnetisation obtained from hysteresis loops of the investigated samples at T = 5 K
EPR spectra of five investigated samples at three different temperatures (90, 190, and 290 K) are presented in Fig. 10. In general, the spectra have small (samples S20, S23) or very small (S19, S21, S22) amplitude and are more pronounced at RT than at lower temperatures. They consist of very broad lines with anisotropic shape. To discuss the temperature dependence of EPR spectra, each spectrum was fitted with one or more lorentzian/gaussian components. This rough treatment of the registered spectra was used to obtain only very general information on their thermal behaviour. Figure 11 presents, as an example, such fits of EPR spectra of samples registered at T = 90 K. To obtain satisfactory fits to experimental spectra, only one component was sufficient for S19, S21, and S22 samples (lorentzian-type lineshape), but two components were needed for S20 and S23 samples. In the latter case, the main component has lorentzian-type lineshape, while the additional component with gaussian-type lineshape was the best to fit experimental spectra. Fitting enabled calculation of the three spectral parameters—the resonance field Br, peak-to-peak linewidth ΔB, and integrated intensity Iint. The latter is calculated as the product of the amplitude A and square of the linewidth, Iint = A·ΔB2. It is proportional to the magnetic susceptibility of the spin system involved in the formation of EPR spectrum at microwave frequency as well as to the concentration of the EPR active centers.

EPR spectra of samples S19, S20, S21 (a) and S22, S23 (b) recorded at T = 90, 190, and 290 K

Experimental (symbols) and fitted (lines) EPR spectra of samples S19, S20, S21 (a) and S22, S23 (b) registered at T = 90 K
Figure 12 shows the temperature dependence of the resonance field (a) and peak-to-peak linewidth (b) of individual components in each sample, while Fig. 13 presents the temperature dependence of the integrated intensity of each separate component (a) and the total (being the sum of all components) integrated intensity (b) in five investigated samples. The temperature dependencies of EPR parameters are quite different for the additional components (S20b and S23b) than for the rest main components (S19, S20a, S21, S22, S23a). For the latter group, the resonance fields are in 2.85–3.00 kG range (equivalent g-factors are in 2.37–2.25 range) at RT and do not change much on cooling samples down to 90 K, the linewidths are in 1.2–1.8 kG range and increase on temperature decrease, and the integrated intensities initially decrease on cooling samples from RT, but below ~ 150 K they start to increase on further cooling. In contrast, for the additional components, the resonance fields are in 4.1–4.2 kG range (g-factor in 1.65–1.61 range) at RT and appear to be independent on temperature, the linewidths are about 1 kG and show a maximum at temperature ~ 150 K, while the integrated intensities display a constant decrease with temperature decrease from RT.

Temperature dependence of the resonance field (a) and peak-to-peak linewidth (b) of the individual components in the EPR spectra

Temperature dependence of the integrated intensity of the individual components (a) and total integrated intensity (b) in the EPR spectra
The main components in all studied samples might be attributed to two spin subsystems—one encompassing AFM spin pairs/cluster with a non-magnetic ground level (S = 0) that is responsible for intensity increase at high temperatures, and the other subsystem of paramagnetic spin cluster that accounts for EPR signal increase at lower temperatures. On the other hand, the additional EPR components (S20b and S23b) might be due to another AFM spin pair/cluster, with different internal fields and different relaxation times in comparison to the previous one. An overall weak temperature dependence of EPR spectrum parameters indicates that the spin–spin interaction (dipole or exchange) plays an important role in the formation of EPR response in our samples.
4 Conclusions
The studied five samples from FeVO4–Co3V2O8 system were diphase, containing L-type phase and Co3V2O8. The content of L-type phase decreased with the increased content of Co in initial mixtures. The magnetic ions Co2+ and Fe3+ were in the high-spin state S = 3/2 and 5/2, respectively. The observed effective magnetic moment inferred from the Curie constant was higher than theoretical one, indicating an additional contribution from the defective V4+ (S = ½) ions. AFM interaction between paramagnetic ions was determined from the negative Curie–Weiss temperature. The strength of this AFM interaction diminished going from sample S19 to S21, but increased on further Co content increase in samples S22 and S23. At low temperatures, two magnetic transitions are found (at T = 6 K and 12 K) that might be related to transitions in Co3V2O8 phase. Magnetic anisotropy deduced from (χFC − χZFC) difference was small and similar in value in all samples with exception of sample S20. This might be associated with bigger sizes of nanometric spin clusters in that sample. A FM component was identified from hysteresis loop in all samples at T = 5 K. In samples S21, S22, and S23, Hc < 100 Oe and Mr < 1.3 emu/g, while in samples S19 and S20 Hc ~ 1000 Oe, Mr > 3 emu/g. These differences might be correlated with a higher content of Co3V2O8 phase in the former samples evidenced by XRD method. EPR spectra were either weak (samples S20, S23) or very weak (samples S19, S21, S22), indicating they were formed by structural and magnetic defects. Very broad lines in EPR spectra in all samples were attributed to nanometric magnetic spin pairs/clusters. In general, samples showed inhomogeneous magnetism due to the presence of two strong magnetic ions randomly distributed between different crystallographic sites and a mixture of two magnetic phases.
References
M.A. Lafontaine, J.M. Greneche, Y. Laligant, G. Ferey, J. Solid State Chem. 108, 1–10 (1994)
B. Zhaorigetu, W. Li, R. Kieffer, H. Xu, React. Kinet. Catal. Lett. 75, 275–287 (2002)
X. Wang, D.A. Vander Griend, ChL Stern, K.R. Poeppelmeier, Inorg. Chem. 39, 136–140 (2000)
A.A. Belik, F. Izumi, T. Ikeda, A. Nisawa, T. Kamiyama, K. Oikawa, Solid State Sci. 4, 515–522 (2002)
A. Blonska-Tabero, M. Kurzawa, J. Therm. Anal. Calorim. 88, 33–36 (2007)
V.G. Krivovichev, M.V. Charykova, Geol. Ore Deposits 59, 677–686 (2017)
Z. He, J.-I. Yamaura, Y. Ueda, J. Solid State Chem. 181, 2346–2349 (2008)
V.D. Nithya, R. Kalai Selvan, C. Sanjeeviraja, D. Mohan Radheep, S. Arumugam, Mater. Res. Bull. 46, 1654–1658 (2011)
A. Dixit, B. Ramchandran, Y.K. Kuo, G. Lawes, IEEE T. Magn. 51, 2504304 (2015)
Y. Chen, J.W. Lynn, Q. Huang, F.M. Woodward, T. Yildirim, G. Lawes, A.P. Ramirez, N. Rogado, R.J. Cava, A. Aharony, O. Entin-Wohlman, A.B. Harris, Phys. Rev. B 74, 014430 (2006)
Z. He, T. Taniyama, M. Itoh, Y. Ueda, Cryst. Growth Des. 7, 1055–1057 (2007)
Y. Yasui, Y. Kobayashi, M. Soda, T. Moyoshi, M. Sato, N. Igawa, K. Kakurai, J. Phys. Soc. Jpn. 76, 034706 (2007)
J.S. Helton, Y. Chen, G.L. Bychkov, S.N. Barilo, N. Rogado, R.J. Cava, J.W. Lynn, J. Phys.: Condens. Matter 24, 016003 (2012)
G. Gao, S. Lu, B. Dong, Y. Xiang, K. Xi, S. Ding, J. Mater. Chem. A 4, 6264–6270 (2016)
B. Sambandam, V. Soundharrajan, V. Mathew, J. Song, S. Kim, J. Jo, D.P. Tung, S. Kim, J. Kim, J. Mater. Chem. A 4, 14605–14613 (2016)
T. Hyeon, Y. Chung, J. Park, S.S. Lee, Y.-W. Kim, B.H. Park, J. Phys. Chem. B 106, 6831–6833 (2002)
N. Guskos, G. Zolnierkiewicz, J. Typek, A. Blonska-Tabero, Phys. B 406, 2163–2167 (2011)
N. Guskos, G. Zolnierkiewicz, J. Typek, R. Szymczak, A. Guskos, P. Berczynski, A. Blonska-Tabero, Mater. Sci. Poland 31, 601–610 (2013)
N. Guskos, G. Zolnierkiewicz, M. Pilarska, J. Typek, A. Blonska-Tabero, C. Aidinis, Acta Phys. Pol. A 132, 24–29 (2017)
N. Guskos, G. Zolnierkiewicz, M. Pilarska, J. Typek, P. Berczynski, A. Blonska-Tabero, C. Aidinis, J. Phys. Chem. Solids 115, 156–161 (2018)
J. Lopez, L.F. Gonzalez-Bahamon, J. Prado, J.C. Caicedo, G. Zambrano, M.E. Gomez, J. Esteve, P. Prieto, J. Magn. Magn. Mater. 324, 394–402 (2012)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Guskos, N., Zolnierkiewicz, G., Pilarska, M. et al. EPR and Magnetometry of Mixed Phases in FeVO4–Co3V2O8 System. Appl Magn Reson 50, 737–751 (2019). https://doi.org/10.1007/s00723-018-1064-4
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-018-1064-4