
Summary.
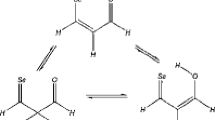
The electronic and geometry structures of all enol and diketoforms of 3-oxopropionyl halogenides were investigated at the B3LYP/3-21G** level of theory. The comparative study of their energies revealed that the enol forms of the compounds are more stable than cis- and trans-diketones. All cis-diketones (except Cl-containing one) have one imaginary frequency in their vibration spectra, i.e. they do not correspond to local or global minima on the potential hypersurface. The keto–enol transformations enol⇆cis-diketone and enol⇆trans-diketone for a certain triad of tautomers/isomers pass through one transition state or in other words, the two conversions have one common maximum on the energy hypersurface. The energy barriers of the keto–enol conversions found for isolated molecules are quite high. Only for the enol → diketone conversions in solution the energy barriers are slightly reduced.
Similar content being viewed by others

Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Delchev, V. A DFT Study of Electron Structure, Geometry, and Keto–Enol Tautomerism of 3-Oxopropionyl Halogenides. Monatshefte für Chemie 135, 371–384 (2004). https://doi.org/10.1007/s00706-003-0132-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-003-0132-z