
Abstract
The family Geminiviridae includes plant-infecting circular single-stranded DNA viruses that have geminate particle morphology. Members of this family infect both monocotyledonous and dicotyledonous plants and have a nearly global distribution. With the advent of new molecular tools and low-cost sequencing, there has been a significant increase in the discovery of new geminiviruses in various cultivated and non-cultivated plants. In this communication, we highlight the establishment of three new genera (Becurtovirus, Eragrovirus and Turncurtovirus) to accommodate various recently discovered geminiviruses that are highly divergent and, in some cases, have unique genome architectures. The genus Becurtovirus has two viral species, Beet curly top Iran virus (28 isolates; leafhopper vector Circulifer haematoceps) and Spinach curly top Arizona virus (1 isolate; unknown vector), whereas the genera Eragrovirus and Turncurtovirus each have a single assigned species: Eragrostis curvula streak virus (6 isolates; unknown vector) and Turnip curly top virus (20 isolates; leafhopper vector Circulifer haematoceps), respectively. Based on analysis of all of the genome sequences available in public databases for each of the three new genera, we provide guidelines and protocols for species and strain classification within these three new genera.
Similar content being viewed by others

Introduction
Geminiviruses are single-stranded DNA viruses with either monopartite or bipartite genomes that are encapsidated within virions that have a geminate morphology. Different geminiviruses can infect either monocotyledonous or dicotyledonous plants, and collectively, these viruses cause a variety of major crop diseases. Many of these diseases pose a serious threat to the food security of developing countries in the tropical and subtropical regions of the world [25]. Due primarily to the development of new molecular tools and the proliferation of industrial-scale low-cost commercial sequencing services (applying both Sanger and a variety of “next-generation” sequencing techniques), there has been a tremendous increase over the last five years in the rate at which new geminivirus genomes have been characterised. Of the new molecular tools, sequence-independent methodologies based on rolling-circle amplification that employ ϕ29 DNA polymerase have played a disproportionately important role in the discovery of new highly divergent geminiviruses [17, 20, 27, 31].
The Geminiviridae Study Group of the International Committee on Taxonomy of Viruses (ICTV) has proposed various guidelines on how new geminivirus genome sequences should be taxonomically classified and named [6, 12, 13, 26, 36]. At the genus level, geminiviruses have been classified based on host range, insect vector, genome organisation and genome-wide pairwise sequence identities, and the Ninth Report of the ICTV [6] acknowledged the existence of four genera: Mastrevirus, Begomovirus, Curtovirus and Topocuvirus.
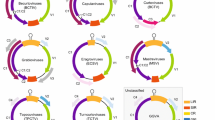
In this communication, we provide an update of the 9th Report with the establishment of three additional genera in the family Geminiviridae to accommodate divergent new groups of geminiviruses that have been discovered and characterised over the last five years (Fig. 1):

Genome organisation of isolates in various geminivirus lineages (LIR, long intergenic region; SIR, short intergenic region; CR, common region; rep, replication-associated protein; ren, replication enhancer; trap, transactivator protein; ss, silencing suppressor; tgs, transcriptional gene silencing; sd, symptom determinant; cp, capsid protein; mp, movement protein; reg, regulatory gene; NSP, nuclear shuttle protein). Please note that the DNA-A components of Old World bipartite begomoviruses contain a V2 ORF. The V2, C2 and C4 ORFs of begomoviruses (TYLCV, TYLCSV and TGMV) have been shown to suppress transcriptional gene silencing [7, 24, 37]
1) Becurtovirus: Including the species Beet curly top Iran virus and Spinach curly top Arizona virus (Table 1) [14, 18, 19, 33, 39]
2) Eragrovirus: Including the species Eragrostis curvula streak virus (Table 2) [35]
3) Turncurtovirus: Including the species Turnip curly top virus (Table 3) [5, 29, 30]
We additionally acknowledge the need for further new genera to accommodate three other highly divergent groups of geminiviruses that have recently been found infecting grapevines in the USA [22, 28] and citrus plants in Turkey [23], as well as two closely related viruses (~72 % genome-wide pairwise sequence identity) infecting Euphorbia caput-medusae in South Africa [4] and French beans in India.
Whereas the genome organisations of begomoviruses, curtoviruses, topocuviruses, and turncurtoviruses are quite similar, those of the mastreviruses, becurtoviruses and eragroviruses, are quite diverse and distinctive from other geminiviruses (Fig. 1). In general, these various geminivirus genomes encode up to eight genes, which are transcribed bidirectionally. Of these, the replication-associated protein gene (rep), located on the complementary-sense strand, and the capsid protein gene (cp), located on the virion-sense, are the only two that are conserved in all characterised geminiviruses. Whereas homologues of the replication enhancer (ren) genes and symptom determinant (sd)/silencing suppressor (ss) genes are found in the genomes of begomoviruses, curtoviruses, topocuviruses and turncurtoviruses, homologues of the transcription activator (trap) gene are additionally found in eragroviruses (Fig. 1).
Interestingly, despite the probable movement protein genes (mp) of all geminiviruses being located in the same region, downstream of the intergenic region (IR) from which transcription begins, these proteins are very diverse, and there is generally no detectable amino acid similarity between the MP sequences of viruses from different genera.
Another genomic feature that distinguishes the mastreviruses, eragroviruses and becurtoviruses from the other geminiviruses is the presence of two IRs: one containing the virion-strand origin of replication (although it is called the long or large IR [LIR] in mastreviruses, this is a misnomer in eragroviruses because it is the smaller of the two intergenic regions) and another containing the transcription termination signals of the bidirectionally transcribed genes (called the small or short IR [SIR] in mastreviruses).
Besides obvious differences in genome architectures, the viruses in the different genera share what are probably evolutionarily conserved gene expression strategies. For example, whereas the mastreviruses and becurtoviruses all potentially express two different replication-associated proteins from alternatively spliced complementary-sense transcripts, the mastreviruses additionally produce alternatively spliced virion-strand transcripts [38] (Fig. 1).
The evolutionary relationships between the members of the different geminivirus genera are difficult to unravel, both because it is very likely that recombination has played a fundamental role in the evolution of these viruses and because large portions of the genomes of viruses from any two different genera are not detectably homologous. With these reservations in mind, we roughly determined the evolutionary relationships between viruses in the different genera (it is important to note that there is a high degree of alignment uncertainty) using MUSCLE [10, 11] to align representative complete genomes of viruses in the different genera (Supplementary Table S1) and the neighbor-joining method (implemented in MEGA 5.2 [34]) with the Jukes-Cantor nucleotide substitution model and 1000 bootstrap replicates to produce a phylogenetic tree (Fig. 2). As suggested by similarities in their genome organisations, the begomoviruses, becurtoviruses, curtoviruses, topocuvirus and turncurtoviruses cluster together, whereas the other groups all form distinct lineages branching directly from the presumed root (in this case, simply the midpoint of the tree between the two most divergent sequences). The only minor exception is that the divergent geminiviruses from E. caput-medusae, French bean and grapevine form a diverse cluster with 70 % bootstrap support.

Neighbor-joining phylogenetic tree of the full genome sequences of representative geminiviruses. This phylogenetic tree is provided only as a guide to describe relative similarities between the geminivirus groups, as (1) it is not possible to credibly align all of the representative genomes from various recognised genera and novel divergent geminivirus isolates, and (2) inter–species and inter-genus recombination has played a major part in the diversification of the represented virus isolates and largely invalidates the use of such a tree to accurately express evolutionary relationships. This tree is midpoint rooted
In order to gain better insights into the evolutionary relationships between the main geminivirus groups, we generated alignments of the Rep and CP amino acid sequences using MUSCLE [10, 11] and inferred maximum-likelihood phylogenetic trees with PHYML [15, 16] using the RtREV+G+I+F model of amino acid substitution (the best-fit model as determined using ProtTest; [1, 8]; Fig. 3) with approximate likelihood ratio test (aLRT) for branch support [3]. Whereas the Rep sequences of becurtoviruses, turncurtoviruses, mastreviruses and eragroviruses are monophyletic, for the CP sequences of these viruses only the begomoviruses and turncurtoviruses are monophyletic. Other important differences between the Rep and CP phylogenies include both the fact that the Rep sequences of becurtoviruses are monophyletic whereas those of their CPs are not, and the fact that the Rep sequences from Caput medusae, French bean and grapevine are monophyletic whereas their CP sequences are not. Among the most obvious causes of these discrepancies are that (1) the CP sequences of becurtoviruses and curtoviruses share >76.5 % pairwise amino acid sequence identity, whereas their Rep sequences share <33 % identity, and (2) the CP sequences of eragroviruses convincingly cluster within the mastrevirus clade and are clearly most closely related to the Eurasian dwarf viruses wheat dwarf virus and oat dwarf virus (Fig. 3), but their Rep sequences are completely distinct from all other known geminiviruses.

Maximum-likelihood (ML) phylogenetic trees of the Rep and CP amino acid sequences of representative geminiviruses. The Rep ML phylogenetic tree has been rooted with Rep sequences of cassava-associated circular DNA virus (JQ412057) [9] and Sclerotinia sclerotiorum hypovirulence-associated virus-1 (GQ365709) [21, 40], whose Rep sequences are distantly related to those of geminiviruses [32]. The CP tree has simply been rooted at the midpoint along the branch path separating the two most divergent sequences represented
These phylogenetic analyses, coupled with obvious differences in the genome organisations of the various divergent geminiviruses analysed here provide clear support for the creation of new genera within the family Geminiviridae to accommodate viruses that cannot justifiably be assigned to the genera Begomovirus, Curtovirus, Mastrevirus, or Topocuvirus. Accordingly, three new geminivirus genera are now recognised by the ICTV [2].
Becurtovirus
This genus contains two recognised species: Beet curly top Iran virus and Spinach curly top Arizona virus (Table 1). Members of both species are unusual in that instead of the “TAATATTAC” nonanucleotide that is found at the origin of virion-strand replication in almost all other known geminiviruses, these have a “TAAGATTCC” nonanucleotide. Isolates belonging to these two species have genomes that share between 77 and 78 % identity. The 28 full-genome sequences of beet curly top Iran virus (BCTIV) that were available in GenBank on 22 October 2013 all share >88.6 % genome-wide pairwise identity and have been found exclusively in Iran where they have been isolated from the dicotyledonous plants of the species Beta vulgaris, Beta vulgaris subsp. maritima, Vigna unguiculata, Solanum lycopersicum and Phaseolus vulgaris [14, 19, 39]. The only SCTAV isolate that has so far been fully sequenced was found infecting Spinacia oleracea plants in Arizona, USA [18]. Genome-wide pairwise identity analysis of the 29 available becurtovirus genomes (404 pairwise comparisons) using SDT v1.0 (with the MUSCLE alignment option; available for download from http://web.cbio.uct.ac.za/SDT [26]) revealed pairwise identity peaks between 76 and 79 %, between 88 and 93 %, and between 95 and 100 % and valleys between 80 and 87 % and between 93 and 95 % (Fig. 4). Based on this distribution of pairwise identities, we propose tentative species and strain demarcation thresholds of 80 % and 94 %, respectively. Hence, pairs of becurtovirus full-genome sequences with >80 % pairwise identity calculated from pairwise alignments produced using the MUSCLE program with default settings (i.e., as it is implemented in the program SDT v1.0 [26]) should for now be considered members of the same species. Similarly, pairs of genomes with >94 % pairwise identity using this same analysis approach should for now be considered as members of the same strain. Please note that both of these criteria are tentative in that they are based on the information available for only two species. As more becurtovirus sequences are catalogued, these thresholds may change.

Distribution of pairwise identities of (a) becurtoviruses (404 pairwise comparisons), (b) eragroviruses (15 pairwise comparisons), and (c) turncurtoviruses (190 pairwise comparisons), calculated using SDT v1.0 [26] with the MUSCLE-alignment option
Under this classification system, the genus includes two species, with one of these, Beet curly top Iran virus, including four strains. As has been done for the mastreviruses, we have opted to simply name these strains alphabetically [26]. This should, however, not be seen as precluding the naming of strains based on consistently observable biological differences between the members of different strains (see Table 1 for the nomenclature).
Eragrovirus
This genus has only one recognised species, Eragrostis curvula streak virus. All known isolates belonging to this species have been found infecting monocotyledonous plants of the species Eragrostis curvula (weeping love grass) in the Kwa-Zulu Natal region of South Africa (Table 2) [35]. As is the case with the becurtoviruses, all six isolates of Eragrostis curvula streak virus (ECSV) have an unusual “TAAGATTCC” nonanucleotide sequence motif at their origin of virion-strand replication. These six ECSV isolates all share >91.6 % genome-wide sequence identity, and the distribution of identities between all 15 unique pairs of these viruses (as determined from MUSCLE alignments produced with default settings using SDT v1.0) reveal peaks between 91 and 93 %, between 94 and 97 %, and between 98 and 100 % and valleys at 93 % and 97 % (Fig. 4). Based on this distribution of pairwise identities, we propose a tentative strain demarcation threshold of 94 % (i.e., the same as that proposed for the becurtoviruses). For now, pairs of eragrovirus full-genome sequences that have >94 % pairwise identity as determined by the method implemented in SDT1.0 should be considered members/variants of the same strain. Based on this criterion, we have classified the six known ECSV sequences into two alphabetically named strains (see Table 2). As is the case with becurtoviruses, our simple alphabetic naming system should not preclude the future naming of strains based on consistently observable biological differences between the members of different strains (see Table 2 for the nomenclature).
Turncurtovirus
Turnip curly top virus is currently the only species within this genus. The 20 isolates belonging to this species that are known so far have all been recovered from either Brassica rapa or Raphanus sativus [5, 30]. Turnip curly top virus (TCTV) has, however, also been detected by PCR in Descurainia sophia, Anchusa sp., Solanum americanum and Hibiscus trionum [29]. All 20 of the known TCTV isolates have the same “TAATATTAC” nonanucleotide sequence motif that is found at the virion-strand origins of replication of mastreviruses, begomoviruses, curtoviruses and topocuviruses. All of these isolates share >86.8 % genome-wide pairwise identity, and the distribution of the 190 pairwise identities of every unique pair of these 20 sequences (calculated as described above using SDT v1.0; [26]) has peaks between 86 and 88 %, between 89 and 91 %, between 93 and 95 %, and between 98 and 100 % and valleys between 88 and 89 %, at 92 %, and between 95 and 98 %. Based on this distribution of pairwise identities, we propose a tentative turncurtovirus strain demarcation threshold of 95 %. For now, pairs of turncurtovirus genomes with >95 % pairwise identity, as calculated using an exactly equivalent approach to that described above (using SDT v1.0), should be considered members/variants of the same strain. Based on this criterion, four alphabetically named TCTV strains are proposed. As with the becurtoviruses and eragroviruses, this simple naming system is not intended to preclude the naming of strains based on consistently observable biological differences between the members of different strains (see Table 3 for the nomenclature).
Virus nomenclature
We suggest that the following system be used for naming new becurtoviruses, eragroviruses and turncurtoviruses:
<virus name> - <strain name> [<country/territory code>-<lab codes/old names/host species of origin/sample number/location of origin>-<year of sampling>]
Bounded by square brackets (“[ ]”) the isolate descriptor field may contain any number of sub-fields, each separated by a hyphen (“-”). The first sub-field should be the two-letter international code of the country/territory from which the isolate was originally sampled, i.e., it should be the country where either the virus itself or the plant carrying the virus was sampled from the field (see Supplementary Table S2 for two-letter country codes). The last sub-field should be the year when viral DNA was last present within living tissue, i.e., it should be the year when the plant tissue sample was picked from a living plant and was either stored (dried or frozen) or used for DNA extraction, and not simply the year when viral genomic DNA extraction was carried out. Between the first and the last fields, any number of useful descriptors can be used, including lab ID, field code, region of sampling and host species.
Step-by-step guide to classifying new becurtoviruses, eragroviruses and turncurtoviruses
1) A ‘nucleotide BLAST’ search (accessible via http://blast.ncbi.nlm.nih.gov/Blast.cgi) of the NCBI ‘Nucleotide’ database should be performed to identify the species whose members have sequences most similar to the new sequence.
-
a.
The nucleotide database at the NCBI website (http://www.ncbi.nlm.nih.gov/nuccore/) can also be searched using the search term “txid1298630[Organism:exp] AND 2500:4000[SLEN]”, which will return all becurtovirus nucleotide sequences that are between 2500 and 4000 nucleotides long.
-
b.
The nucleotide database at the NCBI website (http://www.ncbi.nlm.nih.gov/nuccore/) can also be searched using the search term “txid1298632[Organism:exp] AND 2500:4000[SLEN]”, which will return all eragrovirus nucleotide sequences that are between 2500 and 4000 nucleotides long.
-
c.
The nucleotide database at the NCBI website (http://www.ncbi.nlm.nih.gov/nuccore/) can also be searched using the search term “txid1298631 [Organism:exp] AND 2500:4000[SLEN]”, which will return all turncurtovirus nucleotide sequences that are between 2500 and 4000 nucleotides long.
2) The new sequence should be added to the set of sequences (becurtoviruses, eragroviruses or turncurtoviruses) obtained from the NCBI BLAST or the NCBI nucleotide database websites and should be saved in FASTA format. We have provided datasets of all full genomes of becurtoviruses, eragroviruses and turncurtoviruses available in GenBank as of 22 October 2013 (Supplementary Files S1, S2 and S3).
3) Regardless of how datasets are compiled, sub-genome-length sequences should not be used in pairwise sequence identity analyses and hence should be removed from FASTA files prior to such analyses.
4) Before analyses are carried out, it should be ensured that all of the sequences being analysed start at the same genomic coordinate (ideally at the nicking site within the conserved nonanucleotide at the origin of replication).
5) Either the MUSCLE option in SDT v1.0 (available at http://web.cbio.uct.ac.za/SDT) or an exactly equivalent method should be used to calculate identities between every pair of sequences in the dataset. These pairwise identities should be saved in SDT v1.0 in either a column or matrix csv format. The csv file thus produced can then be opened in a spreadsheet program such as Microsoft Excel.
6) In the case of becurtoviruses, if the sequence shares <80 % genome-wide pairwise identity to any other known becurtovirus sequence, an appropriate species name should be proposed. If the sequence is classified as belonging to a particular species but shares <94 % genome-wide pairwise identity to all other known isolates belonging to that species, either a unique alphabetical letter should assigned as a strain name or an alternative strain name may be chosen based on a phenotypic characteristic that distinguishes the isolate from all others that are known.
7) In the case of eragroviruses, if the sequence of a new isolate shares <75 % sequence identity with any other described eragrovirus, it should tentatively be assigned to a new species. Note that this recommendation is simply intended as a conservative guide and that as more eragroviruses are discovered it will likely be changed. If the sequence of a new isolate shares <94 % genome-wide pairwise identity to all other known eragrovirus isolates, it should be assigned to a new strain, which should be named either with a unique letter or according to some defining phenotypic characteristic.
8) In case of turncurtoviruses, if the sequence of a new isolate shares <75 % sequence identity with any other described turncurtovirus it should tentatively be assigned to a new species. Note that this recommendation is simply intended as a conservative guide and that as more turncurtoviruses are discovered it could be changed. If the sequence of a new turncurtovirus isolate shares <95 % genome-wide pairwise identity to all other known turncurtovirus isolates then it should be assigned to a new strain which should be named either with a unique letter or according to some defining phenotypic characteristic of that isolate.
Conclusion
In this communication, we update the list of genera within the family Geminiviridae to accommodate three groups of highly divergent geminiviruses. Among these new genera, the genus Becurtovirus has two recognised species, Beet curly top Iran virus and Spinach curly top Arizona virus, with BCTIV isolates subdivided into four strains (BCTIV-A, -B, -C and -D), the genus Eragrovirus has one recognised species, Eragrostis curvula streak virus, with two strains (ECSV-A and-B), and the genus Turncurtovirus has one recognised species, Turnip curly top virus, with four strains (TCTV-A, -B, -C and -D). We have also provided protocols for the establishment of species and strains within these genera. Although the tentative strain and species demarcation criteria that we have chosen would need to be refined as more sequences are assigned to these genera, we hope that they will provide some uniformity with respect to the classification of newly determined sequences. The Executive Committee of the ICTV has approved the establishment of these three new genera within the family Geminiviridae [2].
References
Abascal F, Zardoya R, Posada D (2005) ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105
Adams MJ, King AM, Carstens EB (2013) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2013). Arch Virol 158:2023–2030
Anisimova M, Gascuel O (2006) Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst Biol 55:539–552
Bernardo P, Golden M, Akram M, Nadarajan N, Naimuddin, Fernandez E, Granier M, Rebelo AG, Peterschmitt M, Martin DP, Roumagnac P (2013) Identification and characterisation of a highly divergent geminivirus: evolutionary and taxonomic implications. Virus Res 177:35–45
Briddon RW, Heydarnejad J, Khosrowfar F, Massumi H, Martin DP, Varsani A (2010) Turnip curly top virus, a highly divergent geminivirus infecting turnip in Iran. Virus Res 152:169–175
Brown JK, Fauquet CM, Briddon RW, Zerbini M, Moriones E, Navas-Castillo J (2012) Virus taxonomy: Ninth report of the International Committee on Taxonomy of Viruses. Academic press, London
Buchmann RC, Asad S, Wolf JN, Mohannath G, Bisaro DM (2009) Geminivirus AL2 and L2 proteins suppress transcriptional gene silencing and cause genome-wide reductions in cytosine methylation. J Virol 83:5005–5013
Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165
Dayaram A, Opong A, Jaschke A, Hadfield J, Baschiera M, Dobson RC, Offei SK, Shepherd DN, Martin DP, Varsani A (2012) Molecular characterisation of a novel cassava associated circular ssDNA virus. Virus Res 166:130–135
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform 5:113
Fauquet CM, Maxwell DP, Gronenborn B, Stanley J (2000) Revised proposal for naming geminiviruses. Arch Virol 145:1743–1761
Fauquet CM, Briddon RW, Brown JK, Moriones E, Stanley J, Zerbini M, Zhou X (2008) Geminivirus strain demarcation and nomenclature. Arch Virol 153:783–821
Gharouni Kardani S, Heydarnejad J, Zakiaghl M, Mehrvar M, Kraberger S, Varsani A (2013) Diversity of beet curly top Iran virus isolated from different hosts in Iran. Virus Genes 46:571–575
Guindon S, Delsuc F, Dufayard JF, Gascuel O (2009) Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol 537:113–137
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Haible D, Kober S, Jeske H (2006) Rolling circle amplification revolutionizes diagnosis and genomics of geminiviruses. J Virol Methods 135:9–16
Hernandez-Zepeda C, Varsani A, Brown JK (2013) Intergeneric recombination between a new, spinach-infecting curtovirus and a new geminivirus belonging to the genus Becurtovirus: first New World exemplar. Arch Virol 158:2245–2254
Heydarnejad J, Keyvani N, Razavinejad S, Massumi H, Varsani A (2013) Fulfilling Koch’s postulates for beet curly top Iran virus and proposal for consideration of new genus in the family Geminiviridae. Arch Virol 158:435–443
Inoue-Nagata AK, Albuquerque LC, Rocha WB, Nagata T (2004) A simple method for cloning the complete begomovirus genome using the bacteriophage phi29 DNA polymerase. J Virol Methods 116:209–211
Kraberger S, Stainton D, Dayaram A, Zawar-Reza P, Gomez C, Harding JS, Varsani A (2013) Discovery of Sclerotinia sclerotiorum Hypovirulence-Associated Virus-1 in Urban River sediments of heathcote and Styx Rivers in Christchurch City, New Zealand. Genome Announc 1(4):e00559–e005913
Krenz B, Thompson JR, Fuchs M, Perry KL (2012) Complete genome sequence of a new circular DNA virus from grapevine. J Virol 86:7715
Loconsole G, Saldarelli P, Doddapaneni H, Savino V, Martelli GP, Saponari M (2012) Identification of a single-stranded DNA virus associated with citrus chlorotic dwarf disease, a new member in the family Geminiviridae. Virology 432:162–172
Luna AP, Morilla G, Voinnet O, Bejarano ER (2012) Functional analysis of gene-silencing suppressors from tomato yellow leaf curl disease viruses. Mol Plant Microbe Interact 25:1294–1306
Moffat AS (1999) Plant pathology—geminiviruses emerge as serious crop threat. Science 286:1835
Muhire B, Martin DP, Brown JK, Navas-Castillo J, Moriones E, Zerbini FM, Rivera-Bustamante R, Malathi VG, Briddon RW, Varsani A (2013) A genome-wide pairwise-identity-based proposal for the classification of viruses in the genus Mastrevirus (family Geminiviridae). Arch Virol 158:1411–1424
Owor BE, Shepherd DN, Taylor NJ, Edema R, Monjane AL, Thomson JA, Martin DP, Varsani A (2007) Successful application of FTA((R)) Classic Card technology and use of bacteriophage phi 29 DNA polymerase for large-scale field sampling and cloning of complete maize streak virus genomes. J Virol Methods 140:100–105
Poojari S, Alabi OJ, Fofanov VY, Naidu RA (2013) A leafhopper-transmissible DNA virus with novel evolutionary lineage in the family geminiviridae implicated in grapevine redleaf disease by next-generation sequencing. PloS one 8:e64194
Razavinejad S, Heydarnejad J (2013) Transmission and natural hosts of Turnip curly top virus. Iran J Plant Pathol 49:27–28
Razavinejad S, Heydarnejad J, Kamali M, Massumi H, Kraberger S, Varsani A (2013) Genetic diversity and host range studies of turnip curly top virus. Virus Genes 46:345–353
Shepherd DN, Martin DP, Lefeuvre P, Monjane AL, Owor BE, Rybicki EP, Varsani A (2008) A protocol for the rapid isolation of full geminivirus genomes from dried plant tissue. J Virol Methods 149:97–102
Sikorski A, Arguello-Astorga GR, Dayaram A, Dobson RC, Varsani A (2013) Discovery of a novel circular single-stranded DNA virus from porcine faeces. Arch Virol 158:283–289
Soleimani R, Matic S, Taheri H, Behjatnia SAA, Vecchiati M, Izadpanah K, Accotto GP (2013) The unconventional geminivirus Beet curly top Iran virus: satisfying Koch’s postulates and determining vector and host range. Ann Appl Biol 162:174–181
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Varsani A, Shepherd DN, Dent K, Monjane AL, Rybicki EP, Martin DP (2009) A highly divergent South African geminivirus species illuminates the ancient evolutionary history of this family. Virol J 6:36
Varsani A, Martin DP, Navas-Castillo J, Moriones E, Hernandez-Zepeda C, Idris A, Murilo Zerbini F, Brown JK (2014) Revisiting the classification of curtoviruses based on genome-wide pairwise identity. Arch Virol. doi:10.1007/s00705-014-1982-x
Wang B, Li F, Huang C, Yang X, Qian Y, Xie Y, Zhou X (2014) V2 of tomato yellow leaf curl virus can suppress methylation-mediated transcriptional gene silencing in plants. J Gen Virol 95:225–230
Wright EA, Heckel T, Groenendijk J, Davies JW, Boulton MI (1997) Splicing features in maize streak virus virion- and complementary-sense gene expression. Plant J 12:1285–1297
Yazdi HR, Heydarnejad J, Massumi H (2008) Genome characterization and genetic diversity of beet curly top Iran virus: a geminivirus with a novel nonanucleotide. Virus Genes 36:539–545
Yu X, Li B, Fu YP, Jiang DH, Ghabrial SA, Li GQ, Peng YL, Xie JT, Cheng JS, Huang JB, Yi XH (2010) A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. In: Proceedings of the National Academy of Sciences of the United States of America, vol 107. pp 8387–8392
Acknowledgments
AV and DPM are supported by the National Research Foundation, South Africa. JNC and EM are members of the Research Group AGR-214, partially funded by Consejería de Economía, Innovación y Ciencia, Junta de Andalucía, Spain, cofinanced by FEDER-FSE.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Varsani, A., Navas-Castillo, J., Moriones, E. et al. Establishment of three new genera in the family Geminiviridae: Becurtovirus, Eragrovirus and Turncurtovirus . Arch Virol 159, 2193–2203 (2014). https://doi.org/10.1007/s00705-014-2050-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-014-2050-2