
Abstract
Endogenous members of the family Caulimoviridae have now been found in the genomes of many plant species. Although these sequences are usually fragmented and rearranged and show varying degrees of decay, the genomes of the ancestral viruses can often be reassembled in silico, allowing classification within the existing viral taxonomic framework. In this paper, we describe analyses of endogenous members of the family Caulimoviridae in the genomes of Oryza sativa, Nicotiana tabacum and Solanum spp. and on the basis of phylogeny, genome organization and genetic distance within the pol gene, propose two new virus genera called Orendovirus and Solendovirus. A system of nomenclature for endogenous virus sequences in plants is also proposed.
Similar content being viewed by others


Retroelements are genetic entities that occur as both RNA and DNA molecules and alternate between the two through cycles of reverse transcription and transcription. The diversity of viral retroelements is very large and includes retroviruses (family Retroviridae), pararetroviruses (families Caulimoviridae and Hepadnaviridae) and long terminal repeat (LTR) retrotransposons (families Metaviridae and Pseudoviridae) [10, 18]. All viral retroelements contain a gag-pol replicon core, to which is linked additional adaptive genes that enable the different types of retroelement to occupy various ecological niches [17]. The gag gene encodes the major structural protein or capsid protein, and the pol gene encodes an aspartic protease and reverse transcriptase (RT) with RNase H1 activity [10]. The RT has conserved amino acid motifs indicating a common evolutionary origin. Phylogenetic analyses using this part of the protein have led to the development of a universal classification system for retroelements and enabled accurate taxonomic placement, even in the absence of a complete genome sequence [5, 10, 45].
The term ‘endogenous’ is associated with viral retroelements that have infected host germ line cells at some time in the past and are inherited from parent to offspring as provirus in a Mendelian fashion. Within the animal kingdom, endogenous viral retroelements include retroviruses in the genera Alpharetrovirus, Betaretrovirus, Gammaretrovirus and Lentivirus (subfamily Orthoretrovirinae) and LTR retrotransposons in the families Metaviridae and Pseudoviridae [10, 21]. A shared feature of these endogenous viral retroelements is that following integration in the host genome, they evolve in the manner of a pseudogene and accumulate inactivating mutations (premature stop codons, frameshift mutations, gene deletions and internal recombinations) [1, 39]. The extent of sequence decay is dependent on the age of the integration event, and some recently integrated endogenous retroviruses are still relatively intact, transcriptionally active in germ cell tissue and probably infectious [2–4, 12].
Representatives of both the Metaviridae and Pseudoviridae but not the Retroviridae are present in plant genomes [10]. Additionally, a growing number of endogenous members of the family Caulimoviridae have also been identified [14, 28, 35]. Unlike retroviruses and LTR retrotransposons, which encode an integrase, these endogenous members of the Caulimoviridae have inserted in the host genome through other mechanisms, possibly by recruitment of the viral DNA to repair double-stranded breakages in host chromosomal DNA or by recombination of viral pregenomic RNA with LTR retrotransposon RNA to form a chimeric molecule, which then has integrated following normal retrotransposon mechanisms [14, 22].
As with the endogenous members of the family Retroviridae, the majority of endogenous members of the Caulimoviridae appear to be inactive through a variety of mutations. However, there is strong evidence that some sequences are able to initiate infection under certain conditions, the best studied examples being Banana streak OL virus (BSOLV) [27], Banana streak GF virus (BSGfV) [14], Petunia vein clearing virus (PVCV) [33] and Tobacco vein clearing virus (TVCV) [25]. Ageing of the approximate time of integration is possible based on the distribution of similar sequences in related plant species. For example, there are different endogenous badnavirus sequences (<85% nucleotide identity in the pol gene) in Musa acuminata and M. balbisiana [15], progenitors of the domesticated banana, suggesting that the integration events occurred ≤4.6 million years ago, the time at which these two plant species diverged from a common ancestor [23].
Almost all of the endogenous members of the Caulimoviridae remain unclassified, and agreement has not yet been reached on a system of nomenclature. Staginnus et al. [37] recently proposed a system of nomenclature, but this system differentiates between replication-competent and defective endogenous viral sequences. For endogenous sequences with exogenous counterparts, the naming conventions of the International Committee on Taxonomy of Viruses (ICTV) are followed, but for replication-defective endogenous sequences, use of an acronym is proposed, comprising the host plant initials followed by the suffix EPRS (derived from “endogenous pararetroviral sequence”), e.g. SotuEPRS for the endogenous members of the Caulimoviridae in Solanum tuberosum (potato). The latter system of nomenclature is taxonomically imprecise, as ‘pararetrovirus’ is not a recognized taxon name but instead a descriptive term for viruses such as Hepatitis B virus and Cauliflower mosaic virus (CaMV), which use reverse transcription in replication but do not integrate in the host genome as part of the replication cycle [42]. Furthermore, this proposed system of nomenclature is inconsistent with that adopted for the endogenous members of the Retroviridae, which are named in the same manner, irrespective of their replication competency [10].
In this paper, we describe analyses done to classify the endogenous members of the Caulimoviridae for which complete or substantial portions of the genome sequence are available. We do not distinguish between replication-competent and defective sequences, recognizing that a taxon name is an abstract concept and can be applied to both living and extinct organisms. Assignment of a species name to an endogenous sequence does not imply that the sequence is infective, but refers to an ancestral virus, of which the integrated DNA in a plant genome is considered a derivative. An analogy is the fossilized skeleton of an animal or plant embedded in rock, which is mineralized, incomplete and with varying degrees of rearrangement and erosion, but can still be used as evidence by a paleontologist to propose a name and classification. Even though many endogenous members of the Caulimoviridae are probably now replication defective, long stretches of conceptually translatable sequence and even open reading frames often remain, and the phylogenetic signals are typically very strong [15, 19, 22, 36], allowing classification within the existing viral taxonomic framework.
To investigate natural groupings among the endogenous members of the Caulimoviridae, genome organizations and phylogenetic relationships were investigated. Because of incompleteness, the many small (c. 500 nt) badnavirus-like sequences that have been PCR-amplified from members of Musa and other plant genera were excluded from the analyses. Protein sequences corresponding to a region of the CaMV polymerase (pol) from amino acid residues L269 to R672 (GenBank accession NP_056728) were aligned using CLUSTALX [43]. This region includes the seven conserved motifs that define the catalytic region of the reverse transcriptase [29, 45] and conserved residues found in the active site of the RNase H [20, 34]. The protein alignment was then used to write the DNA alignment using the program TRANALIGN, a re-implementation of the program MRTRANS in the EMBOSS suite of software [32]. Endogenous viral sequences that could not be conceptually translated because of mutations were then added to the alignment and selectively realigned using CLUSTALX. Pairwise sequence comparison (PASC) analyses were done using MEGA version 4 [41]. Corrected nucleotide (d(nt)) and amino acid (d(aa)) distances were calculated using the Kimura 2-parameter and Poisson methods, respectively. Phylogenetic analyses were done using several methods, as described below.
Maximum-parsimony analyses were done using PAUP* version 4.0b10 [40]. All substitutions were weighted equally, and gaps were treated as missing data. A heuristic search strategy was implemented with 1,000 replicates using random taxon addition sequence, tree bisection and reconnection (TBR) branch swapping, and a maximum of 50,000 trees per replicate. To assess statistical support, bootstrap support [11] was determined with 1,000 replicates using heuristic search options and TBR branch swapping, with the maxtree option set at 10,000. Bremer support/decay indices [6] were calculated by searching for all trees equal to or less than a given length. A strict consensus of the resulting trees was examined to see which clades were retained. This method gives the minimum number of steps needed to find trees in which a particular clade disintegrates; e.g. clades that are not retained after searching for trees one step longer than the most parsimonious are assigned a value of D1.
Maximum-likelihood analyses were done using the heuristic search strategy in PAUP* version 4.0b10 [40]. ModelTest version 3.07 [31] was used to determine the best-fitting model of DNA substitution for use in maximum-likelihood analyses for each region by selecting a model based on the Akaike Information Criterion. The dataset was not partitioned into RT and RNase H regions, and indels were not included in the maximum-likelihood analyses as implemented in PAUP*, because separate models of evolution cannot be specified for different regions or indels.
Bayesian inference was done, with and without indels and with and without partitioning the RT and RNase H domains, using the program MrBayes v3.1.2 [16]. The RNase H domain was defined as the region spanning amino acid residues P542 to R672 of the CaMV pol based on CLUSTALX alignment to the RNase H domains of the pol of Human immunodeficiency virus (GenBank accession NP_789740), Murine leukemia virus (GenBank accession 2HB5_A) and Rous sarcoma virus (GenBank accession NP_056886) [8, 24]. The analyses used uniform prior distributions for the alpha shape parameter of the gamma distribution (0–10), proportion of invariable sites (0–1), rate matrix parameters (1–100), and branch lengths (1–10). A Dirichlet distribution (four parameters) was used for the base frequencies. Indels were coded as a separate data partition. Each dataset was analysed using the Markov Chain Monte Carlo (MCMC) process starting with an initial random tree, five million generations and four chains. For all analyses, 50,000 trees were sampled from the posterior probability distribution (one every 100 generations), and 10% of the trees were discarded as “burn-in”. A 95% majority-rule consensus tree was calculated in PAUP* for the remaining trees, which served to estimate the posterior probability for each of the resolved clades.
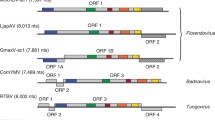
Kunii et al. [22] described a new class of virus-like sequences in the rice (Oryza sativa) genome, which they called endogenous rice tungro bacilliform virus-like sequences (ERTBVs): these sequences formed three distinct clusters, designated A, B and C. The reassembled genomes of these sequences were similar to those of Rice tungro bacilliform virus (RTBV) but differed through the absence of an ORF equivalent to ORF2 in members of the genera Tungrovirus and Badnavirus (Fig. 1a) [22, 38]. Caution must be exercised when making extrapolations about the genome organization of an ancestral virus from an integrated sequence, as an ORF may be missing, either because it is disrupted or because it has been deleted during recombination, but closer examination of the rice sequences suggests that these hypotheses are unlikely. Firstly, homologues of the RTBV ORF2 protein were not retrievable in a tBLASTn search of the O. sativa nucleotide database translated in all six frames, in contrast to the results obtained when a search was done using the RTBV ORF1 protein. Secondly, the structure of the junction between ORF1 and ORF2 is identical for sequence clusters A, B and C, with ORF2 being in a −1 translational reading frame relative to ORF1 and overlapping the end of ORF1 by 34 nucleotides. The junctions of ORFs 1 and 2 and ORFs 2 and 3 in members of the genera Tungrovirus and Badnavirus bear similarity in that each successive ORF is in a −1 reading frame relative to the previous ORF but differ in that only the stop and start codons of successive ORFs overlap.

Comparison of the genome organisation of a Oryza sativa virus (OsatV; GenBank accession BR000031), Rice tungro bacilliform virus (RTBV; GenBank accession NC_001914) and Commelina yellow mottle virus (ComYMV; GenBank accession NC_001343) and b Tobacco vein clearing virus (TVCV; GenBank accession NC_003378) and Cassava vein mosaic virus (CsVMV; GenBank accession NC_001648). Genome maps are linearised, and following convention, numbering begins at the first nucleotide of the tRNAmet binding site. However, this motif could not be found in the intergenic region of OsatV, and the arbritrary start point was designated as T510 in GenBank accession BR000031 based on an optimal alignment of the intergenic regions of OsatV and RTBV. Similarly, parts of the CsVMV ORF1 orthologous to TVCV ORFs 1 and 2 were also determined by alignment of the deduced protein sequences. The TVCV ORF2 protein was first aligned, and then the ORF1 protein to the remaining, truncated sequence of the CsVMV ORF1 protein. Based on this analysis, the CsVMV coat protein extends from M125 (nt 402) to Y875 (nt 2654), and the movement protein from N889 (nt 2694) to K1355 (nt 4094). Conserved motifs are marked with the following symbols and correspond to sequences provided in Fig. 3: movement protein (black club suit), zinc finger (asterisk), aspartic protease active site (open diamonds) and reverse transcriptase active site (shaded diamonds). Dotted lines mark homologous parts of the different virus genomes. Protein molecular weights (kDa) are provided under each ORF label. Arrows denote untranslated regions
All phylogenetic analyses (parsimony, maximum likelihood and Bayesian inference) resulted in similar tree topologies (Fig. 2a, b). There was strong support for a clade containing the monophyletic genera Tungrovirus and Badnavirus and the rice endogenous viruses (Table 1). The strict consensus of all equally parsimonious trees obtained with and without incorporating indels resulted in the endogenous viruses in rice forming a sister clade to the genus Badnavirus, and these two clades in turn were sister to the genus Tungrovirus. In some of the Bayesian analyses, the rice endogenous viruses and the genus Tungrovirus formed a monophyletic clade that was sister to the genus Badnavirus, but this relationship was not retrieved in all Bayesian phylograms obtained. A classification that included the rice endogenous viruses in either of the genera Badnavirus or Tungrovirus would therefore lead to the creation of a paraphyletic group (Fig. 3).

Phylogenetic trees with statistical support using different methods: a Parsimony analysis without indels: strict consensus of 23 equally parsimonious trees (Consistency Index 0.2923, Retention Index 0.5636, Length 10,389) with bootstrap percentages above nodes and Decay Indices (D) below key nodes; b Bayesian inference of sequence data partitioned into RT-RNase H domains, with indels: a randomly selected phylogram, with posterior probabilities above nodes. Abbreviations and sources of sequences are: Saccharomyces cerevisiae Ty3 virus (SceTy3V; GenBank accession M34549), Drosophila melanogaster Gypsy virus (DmeGypV; M12927), Petunia vein clearing virus (PVCV; GenBank accession NC_001839), Soybean chlorotic mottle virus (SbCMV; GenBank accession NC_001739), Blueberry red ringspot virus (BRRV; GenBank accession NC_003138), Peanut chlorotic streak virus (PCSV; GenBank accession NC_001634), Strawberry vein banding virus (SVBV; GenBank accession NC_001725), Cauliflower mosaic virus (CaMV; GenBank accession NC_001497), Carnation etched ring virus (CERV; GenBank accession NC_003498), Horseradish latent virus (HRLV; GenBank accession AY534732), Banana streak OL virus (BSOLV; GenBank accession NC_003381), Commelina yellow mottle virus (ComYMV; GenBank accession NC_001343), Cacao swollen shoot virus (CSSV; GenBank accession NC_001574), Citrus yellow mosaic virus (CiYMV; GenBank accession NC_003382), Dioscorea bacilliform virus (DBV; GenBank accessions X94576 and X94581), Sugarcane bacilliform Mor virus (SCBMorV; GenBank accession NC_008017), Sugarcane bacilliform IM virus (SCBIMV; GenBank accession NC_003031), Banana streak Mys virus (BSMysV; GenBank accession NC_006955), Banana streak OL virus (BSOLV; GenBank accession NC_003381), Kalanchoe top-spotting virus (KTSV; GenBank accession NC_004540), Banana streak GF virus (BSGFV; GenBank accession NC_007002), Oryza sativa virus sequence cluster A (OsatV-A; GenBank accession BR000029), Oryza sativa virus sequence cluster B (OsatV-B; GenBank accession BR000030), Oryza sativa virus sequence cluster C (OsatV-C; GenBank accession BR000031), Rice tungro bacilliform virus isolate Philippines (RTBV-Ph; GenBank accession NC_001914), Rice tungro bacilliform virus isolate West Bengal (RTBV-WB; GenBank accession AJ314596), Cassava vein mosaic virus (CsVMV; GenBank accession NC_001648), LycEPRV-Lh1, -Lh4, -Lh5, -Lh6, -Lh7, -Lh8, -Le1 and -Le2 (GenBank accessions DQ273256, DQ273259, DQ273260, DQ273261, DQ273264, DQ273262, DQ273251 and DQ273252), NtEPRV (GenBank accession AJ238747), NtEPRV-V3, -V4, -V6, -V9, -V14, -E21 and -E22 (GenBank accessions AJ414164, AJ414166, AJ413172, AJ414168, AJ414167, AJ414172 and AJ414173, respectively)

Comparison of conserved motifs [9, 26, 44] in the proteins of a selection of viruses from the genera Badnavirus, Tungrovirus and Cavemovirus and endogenous members of the family Caulimoviridae in the genomes of Oryza sativa, Solanum lycopersicum and Solanum habrochaites. Virus acronyms and sequences are as provided in the caption for Fig. 2. The sequences of LycEPRV-Lh7 and -Lh9 are incomplete, and therefore positions of the motifs in the genome are not shown
Finally, when PASC analyses were done using the pol gene, sequence differences between the rice endogenous viruses and their next closest relatives were similar in magnitude to those of members of different virus genera (Table 2). These results, combined with phylogenetic placement and genome organization, suggest that the ancestor of the rice endogenous viruses should be classified in a new genus within the family Caulimoviridae, for which we propose the name ‘Orendovirus’ (siglum for Oryza endogenous virus). The maximum d(nt) between the endogenous rice virus sequences was 0.107 substitutions per site (9.8% nucleotide identity), suggesting that sequence clusters A, B and C are derived from the same species [5, 10], for which we propose the name Oryza sativa virus (OsatV).
Gambley et al. [13] has found an even closer relative of the rice endogenous viruses in pineapple (Ananas comosus) than either the badnaviruses or tungroviruses, but because of the incompleteness of this sequence, it is not yet possible to tell whether it shares the same genome organization. We propose the name Ananas comosus virus (AcomV) for this virus, but until more is known about its genome organization, we have declined to assign it to a genus within the Caulimoviridae.
Apart from the badnaviruses and the abovementioned rice and pineapple viruses, the other major group of endogenous members of the Caulimoviridae is present in the Solanaceae [19, 25, 36]. All sequences (NtEPRVs and LycEPRVs) share the same genome organization as TVCV [19, 36], and group with this virus in phylogenetic analyses (Fig. 2). The maximum d(nt) between TVCV and these endogenous viruses was 0.222 substitutions per site (19.1% nucleotide identity), and the mean d(nt) was 0.187 substitutions per site (16.4% nucleotide identity). Applying the current threshold for differentiation of species in the family Caulimoviridae (20% nucleotide sequence identity in the pol gene) [10], all sequences derive from the single species, and following naming precedence (chronological order of acceptance by the ICTV), the name Tobacco vein clearing virus should be adopted.
TVCV is currently classified in the genus Cavemovirus, which has Cassava vein mosaic virus (CsVMV) as the type species. In our phylogenetic analyses, there was strong statistical support for CsVMV being sister to the clade containing TVCV and related endogenous virus sequences (Table 1). However, CsVMV has a different genome organisation to TVCV, the key differences being (1) the coat protein and movement protein domains of the ORF1 protein of CsVMV are divided between two ORFs in TVCV; (2) the CsVMV ORF1 protein has a 124-aa N-terminal extension (calculated to be 200 aa by Lockhart et al. [25]) of unknown function relative to the TVCV ORF1 protein and (3) CsVMV has an additional small ORF (ORF2, Fig. 1b) [7, 9, 25]. A fifth ORF described by de Kochko et al. [9], located at nts 7973–8136 in the genome, is unlikely to be functional, as it is within the part of the pregenomic RNA with strong secondary structure that is typically bypassed during translation [30].
PASC analyses of the pol gene by Bousalem et al. [5] suggested that the d(aa) between CsVMV and TVCV is within the range of different virus genera but the d(nt) is more typical of different virus species in the same genus. Our results using a larger fragment of the pol gene support these previous analyses, as the d(aa) between CsVMV and TVCV was at the low end of the range for intergeneric comparisons, but the d(nt) was the lowest of all values (Table 2). Bousalem et al. [5] speculated that this discrepancy between d(aa) and d(nt) could be due to sequencing errors, which would have a disproportionate effect on the amino acid sequence. However, we consider this explanation unlikely, as a full-length, infectious clone of the CsVMV genome was independently sequenced by two groups, and only three discrepancies observed, none of which were within the pol gene [9], and several endogenous TVCV clones have been sequenced and included in our analyses. A more likely explanation is that the rate of non-synonymous substitution in one or other virus has been relatively high due to positive selection pressures. In any case, the d(aa) is more biologically significant than the d(nt) because the protein is the functional unit and should take precedence in considerations on the taxonomy of the viruses.
In conclusion, we recommend that on the basis of differences in genome organisation and PASC analyses, TVCV should be split from the genus Cavemovirus, and a new, monotypic genus should be created, for which we propose the name Solendovirus (siglum for Solanaceae endogenous virus).
To differentiate integrated viral DNA from actively replicating virus, we support the recommendation of Staginnus et al. [37] to place the term ‘endogenous’ prior to the virus species name e.g. endogenous tobacco vein clearing virus, or when abbreviated, eTVCV. Where sequence clusters (sc) occur, this information could be conveyed in a suffix in the manner of a strain designation. When referring to a specific locus, the code for the locus could be provided after the sequence cluster designation e.g. eOsatV-scBLocOs01g02380.1 for endogenous Oryza sativa virus sequence cluster B DNA at locus Os01g02380.1 in the genome of Oryza sativa ssp. japonica cv. Nipponbare. When plant genomes have yet to be sequenced, some other numerical code, a BAC address or even a GenBank accession number could be used until the genome sequence is finalised. In some instances, endogenous DNA from the same ancestral virus species may be in two plant species, either because the integration event preceded plant speciation or because there had been two independent integration events. In these instances, an endogenous virus in one plant species may be named after another plant species, but this problem is no different to that encountered in traditional virus nomenclature, where the virus is named after the host in which it is first found. We recommend that the initials of the plant species be included as the first part of the locus code if not already present.
We do not recommend the use of the suffix ‘a’ or ‘d’ for ‘activateable’ or ‘dead’ viral sequences when referring to a specific locus, as suggested by Staginnus et al. [37], for several reasons. Firstly, the sequence and structural arrangement of a locus are not the only factors determining the ‘activateability’ of a locus, but also the genome composition and ploidy of the host and the prevailing environmental conditions. Secondly, sequences from a number of host loci and even from an exogenous virus may recombine to form an infectious virus genome. Finally, although it may be possible to assign infectivity to a particular locus when the occurrence of infection follows a simple inheritance pattern, as is the case for eBSGFV [14], it would be very difficult to do this when there are multiple or closely linked loci with endogenous viral sequences. To communicate whether a virus species occurs in an endogenous form and whether or not it is extant, categories could be provided in the genus description as has already been done for the genera Alpharetrovirus and Gammaretrovirus [10].
References
Baillie GJ, van de Lagemaat LN, Baust C, Mager DL (2004) Multiple groups of endogenous betaretroviruses in mice, rats, and other mammals. J Virol 78:5784–5798
Belshaw R, Pereira V, Katzourakis A, Talbot G, Pačes J, Burt A, Tristem M (2004) Long-term reinfection of the human genome by endogenous retroviruses. Proc Natl Acad Sci USA 101:4894–4899
Belshaw R, Dawson ALA, Woolven-Allen J, Redding J, Burt A, Tristem M (2005) Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): implications for present-day activity. J Virol 79:12507–12514
Boller K, Schonfeld K, Lischer S, Fischer N, Hoffmann A, Kurth R, Tonjes RR (2008) Human endogenous retrovirus HERV-K113 is capable of producing intact viral particles. J Gen Virol 89:567–572
Bousalem M, Douzery E, Seal S (2008) Taxonomy, molecular phylogeny and evolution of plant reverse transcribing viruses (family Caulimoviridae) inferred from full-length genome and reverse transcriptase sequences. Arch Virol 153:1085–1102
Bremer K (1994) Branch support and tree stability. Cladistics-Int J Willi Hennig Soc 10:295–304
Calvert LA, Ospina MD, Shepherd RJ (1995) Characterization of cassava vein mosaic virus: a distinct plant pararetrovirus. J Gen Virol 76:1271–1278
Davies JF 2nd, Hostomska Z, Hostomsky Z, Jordan SR, Matthews DA (1991) Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science 252:88–95
de Kochko A, Verdaguer B, Taylor N, Carcamo R, Beachy RN, Fauquet C (1998) Cassava vein mosaic virus (CsVMV), type species for a new genus of plant double stranded DNA viruses? Arch Virol 143:945–962
Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (2005) Virus taxonomy: classification and nomenclature of viruses. Eighth report of the international committee on taxonomy of viruses. Elsevier Academic Press, San Diego
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Flockerzi A, Ruggieri A, Frank O, Sauter M, Maldener E, Kopper B, Wullich B, Seifarth W, Muller-Lantzsch N, Leib-Mosch C, Meese E, Mayer J (2008) Expression patterns of transcribed human endogenous retrovirus HERV-K(HML-2) loci in human tissues and the need for a HERV Transcriptome project. BMC Genomics 9:354
Gambley CF, Geering ADW, Steele V, Thomas JE (2008) Identification of viral and non-viral reverse transcribing elements in pineapple (Ananas comosus), including members of two new badnavirus species. Arch Virol 153:1599–1604
Gayral P, Noa-Carrazana J-C, Lescot M, Lheureux F, Lockhart BEL, Matsumoto T, Piffanelli P, Iskra-Caruana M-L (2008) A single Banana streak virus integration event in the banana genome as the origin of infectious endogenous pararetrovirus. J Virol 82:6697–6710
Geering ADW, Olszewski NE, Harper G, Lockhart BEL, Hull R, Thomas JE (2005) Banana contains a diverse array of endogenous badnaviruses. J Gen Virol 86:511–520
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Hull R, Covey SN (1995) Retroelements: Propagation and adaptation. Virus Genes 11:105–118
Hull R (2001) Classifying reverse transcribing elements: a proposal and challenge to the ICTV. Arch Virol 146:2255–2261
Jakowitsch J, Mette MF, van der Winden J, Matzke MA, Matzke AJM (1999) Integrated pararetroviral sequences define a unique class of dispersed repetitive DNA in plants. Proc Natl Acad Sci USA 96:13241–13246
Johnson MS, McClure MA, Feng DF, Gray J, Doolittle RF (1986) Computer analysis of retroviral pol genes: assignment of enzymatic functions to specific sequences and homologies with nonviral enzymes. Proc Natl Acad Sci USA 83:7648–7652
Katzourakis A, Tristem M, Pybus OG, Gifford RJ (2007) Discovery and analysis of the first endogenous lentivirus. Proc Natl Acad Sci 104:6261–6265
Kunii M, Kanda M, Nagano H, Uyeda I, Kishima Y, Sano Y (2004) Reconstruction of putative DNA virus from endogenous rice tungro bacilliform virus-like sequences in the rice genome: implications for integration and evolution. BMC Genomics 5:14
Lescot M, Piffanelli P, Ciampi A, Ruiz M, Blanc G, Leebens-Mack J, da Silva F, Santos C, D’Hont A, Garsmeur O, Vilarinhos A, Kanamori H, Matsumoto T, Ronning C, Cheung F, Haas B, Althoff R, Arbogast T, Hine E, Pappas G, Sasaki T, Souza M, Miller R, Glaszmann J-C, Town C (2008) Insights into the Musa genome: syntenic relationships to rice and between Musa species. BMC Genomics 9:58
Lim D, Gregorio GG, Bingman C, Martinez-Hackert E, Hendrickson WA, Goff SP (2006) Crystal structure of the Moloney murine leukemia virus RNase H domain. J Virol 80:8379–8389
Lockhart BE, Menke J, Dahal G, Olszewski NE (2000) Characterization and genomic analysis of tobacco vein clearing virus, a plant pararetrovirus that is transmitted vertically and related to sequences integrated in the host genome. J Gen Virol 81:1579–1585
Melcher U (2000) The ‘30 K’ superfamily of viral movement proteins. J Gen Virol 81:257–266
Ndowora T, Dahal G, LaFleur D, Harper G, Hull R, Olszewski N, Lockhart B (1999) Evidence that badnavirus infection in Musa can originate from integrated sequences. Virology 255:214–220
Pahalawatta V, Druffel K, Pappu H (2008) A new and distinct species in the genus Caulimovirus exists as an endogenous plant pararetroviral sequence in its host, Dahlia variabilis. Virology 376:253–257
Poch O, Sauvaget I, Delarue M, Tordo N (1989) Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. The EMBO Journal 8:3867–3874
Pooggin MM, Fütterer J, Skryabin KG, Hohn T (1999) A short open reading frame terminating in front of a stable hairpin is the conserved feature in pregenomic RNA leaders of plant pararetroviruses. J Gen Virol 80:2217–2228
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Rice P, Longden I, Bleasby A (2000) EMBOSS: The European molecular biology open software suite. Trends Genet 16:276–277
Richert-Pöggeler KR, Noreen F, Schwarzacher T, Harper G, Hohn T (2003) Induction of infectious petunia vein clearing (pararetro) virus from endogenous provirus in petunia. EMBO J 22:4836–4845
Schultz SJ, Champoux JJ (2008) RNase H activity: structure, specificity, and function in reverse transcription. Virus Res 134:86–103
Staginnus C, Richert-Poggeler KR (2006) Endogenous pararetroviruses: two-faced travelers in the plant genome. Trends Plant Sci 11:485–491
Staginnus C, Gregor W, Mette MF, Teo C, Borroto-Fernandez E, Machado ML, Matzke M, Schwarzacher T (2007) Endogenous pararetroviral sequences in tomato (Solanum lycopersicum) and related species. BMC Plant Biol 7:24
Staginnus C, Iskra-Caruana M, Lockhart B, Hohn T, Richert-Pöggeler K (2009) Suggestions for a nomenclature of endogenous pararetroviral sequences in plants. Arch Virol 154:1189–1193
Stavolone L, Herzog E, Leclerc D, Hohn T (2001) Tetramerization is a conserved feature of the virion-associated protein in plant pararetroviruses. J Virol 75:7739–7743
Stoye JP (2001) Endogenous retroviruses: Still active after all these years? Curr Biol 11:R914–R916
Swofford DL (2002) PAUP*. Phylogenetic analysis using parsimony (*and other methods). Sinauer Associates, Sunderland, MA
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Temin HM (1985) Reverse transcription in the eukaryotic genome: retroviruses, pararetroviruses, retrotransposons, and retrotranscripts. Mol Biol Evol 2:455–468
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24:4876–4882
Torruella M, Gordon K, Hohn T (1989) Cauliflower mosaic virus produces an aspartic proteinase to cleave its polyproteins. EMBO J 8:2819–2825
Xiong Y, Eikbush TH (1990) Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J 9:3353–3362
Acknowledgments
We thank John Thomas, Roger Hull and Neil Olszewski for critically reading the manuscript. Pierre-Yves Teycheney is supported by the FEDER programme.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Geering, A.D.W., Scharaschkin, T. & Teycheney, PY. The classification and nomenclature of endogenous viruses of the family Caulimoviridae . Arch Virol 155, 123–131 (2010). https://doi.org/10.1007/s00705-009-0488-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-009-0488-4