
Abstract
REM behavior disorder (RBD) is a parasomnia characterized by REM sleep without atonia, leading to abnormal and potentially injurious behavior during REM sleep. It is considered one of the most specific predictors of neurodegenerative disorders, such as Parkinson’s disease. In this paper, we provide an overview of animal models contributing to our current understanding of REM-associated atonia, and, as a consequence, the pathophysiology of RBD. The generator of REM-associated atonia is located in glutamatergic neurons of the pontine sublaterodorsal nucleus (SLD), as shown in cats, rats and mice. These findings are supported by clinical cases of patients with lesions of the homologous structure in humans. Glutamatergic SLD neurons, presumably in conjunction with others, project to (a) the ventromedial medulla, where they either directly target inhibitory interneurons to alpha motor neurons or are relayed, and (b) the spinal cord directly. At the spinal level, alpha motor neurons are inhibited by GABAergic and glycinergic interneurons. Our current understanding is that lesions of the glutamatergic SLD are the key factor for REM sleep behavior disorder. However, open questions remain, e.g. other features of RBD (such as the typically aggressive dream content) or the frequent progression from idiopathic RBD to neurodegenerative disorders, to name only a few. In order to elucidate these questions, a constant interaction between basic and clinical researchers is required, which might, ultimately, create an early therapeutic window for neurodegenerative disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
RBD is a parasomnia characterized by the sustained or intermittent loss of atonia during rapid eye movement (REM) sleep, resulting in abnormal, disruptive, potentially or actually injurious behavior that cannot be explained by another sleep or seizure disorder and that is typically associated with dream mentation (American Academy of Sleep Medicine 2005).
Nocturnal phenomena during REM sleep typically include abnormal vocalizations, such as speaking, screaming or laughing, as well as motor behavior, e.g. kicking, punching or flailing, resembling an altered, typically aggressive dream content.
RBD shows a high association with neurodegenerative disorders, and it is considered one of the most important, if not the most specific, predictor of alpha-synucleinopathies (Boeve 2010; Postuma et al. 2012).
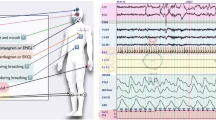
Loss of muscle atonia, and, in addition, active movement generation during REM sleep is the most important and characteristic feature of RBD (Fig. 1). Consequently, a profound understanding of the physiological pathways and processes of REM-associated atonia is crucial in order to gain insight into the potential underlying pathomechanisms. Although basic researchers have been interested in this question for the preceding 20 years, it was certainly further stimulated as a response to the discovery of RBD as a clinical entity by Schenck and colleagues in 1986 (Schenck et al. 1986). In this paper, we provide a concise overview of animal models contributing to our current understanding of REM-associated atonia, which is generated in the sublaterodorsal nucleus (SLD), and modulated at the medullary and spinal level. Currently, the role of the SLD as a core structure for RBD is much better understood than the latter, which remain controversial and will be described less detailed.

Polysomnographic recording of a patient with RBD. During REM sleep, patients with RBD show a loss of physiological muscle atonia, leading to abrupt movements in upper and lower extremities (arrow) and the chin without signs of epileptic activity
The sublaterodorsal nucleus (SLD)
Cortical signs of REM sleep, as well as atonia, are abolished in cats transected at the pontomesencephalic junction, but are present when the pontomedullary junction is severed. In the latter case, however, atonia was defined as “virtual atonia”, i.e. electrophysiological activity in descending pathways. These findings indicate that the generators of REM atonia are located within the pons. The actual site could be further specified, since cats lesioned in the subcoeruleus region, or peri-locus coeruleus (peri-LC) alpha, of the dorsal pontine tegmentum exhibit REM sleep without atonia (Jouvet 1965; Henley and Morrison 1974; Sakai et al. 1979). In the rat, the subcoeruleus region corresponds to the sublaterodorsal nucleus (SLD), a structure slightly rostral to the nucleus subcoeruleus. In support of the abovementioned findings in the cat, REM atonia could be induced in the rat by (1) iontophoretic application of a glutamate agonist, and (2) pharmacological disinhibition by means of microiontophoresis of GABA antagonists into the SLD. The latter could be reversed by local application of kynurenic acid, a glutamate antagonist (Boissard et al. 2002).
The crucial role of the SLD for REM-associated atonia was further demonstrated by Lu, who showed that cytotoxic lesions of the SLD and its immediate surroundings result in REM sleep without atonia including complex movements, such as locomotion (Lu et al. 2006). Both Lu and Luppi demonstrated that SLD neurons with descending spinal projections, which are active during REM-enriched episodes, contain mRNA for vesicular glutamate transporter 2 (VGLUT2), and are therefore glutamatergic. This was further shown by retrograde labeling and Fos studies (Lu et al. 2006) as well as a combined approach using Fos labeling and in situ hybridization (Clement et al. 2011).
The important role of glutamatergic transmission in the SLD for REM-associated atonia was also demonstrated in mice. Using a conditional knockout approach, the expression of vesicular glutamate transporter-2 (Vglut2), and therefore, glutamatergic transmission, was selectively inhibited in the mouse SLD. As a result, the experimental animals could not maintain atonia during REM sleep and exhibited a behavior reminiscent of the phenotype described in Jouvet’s “pontine cat” and lesioned rats, which closely resembled—as much as this is possible to state—human RBD (Krenzer et al. 2011).
In summary, these findings from three different species indicate a crucial role of glutamatergic SLD neurons for the regulation of physiological atonia during REM sleep and the pathophysiology of RBD, as the behavior of all species closely resembled—as far as this is possible to claim—human RBD. Of note, time-lock video recordings are essential to make such a statement, as this is probably the most accurate way of detecting a particular motor behavior in animals. Complex movements, such as locomotion, are an important feature of RBD, and cannot be detected by nuchal EMG alone. If video recordings are not available, extremity EMGs might provide an alternative.
Besides, patients with RBD do not only show behavioral changes in REM sleep, but also an increase in phasic motor activity during non-REM (NREM) sleep, which was not observed in animals with lesions of the SLD. Therefore, additional structural damages to the ventral mesopontine junction (VMPJ), a structure in close proximity to the substantia nigra, might also play a role in RBD (Lai et al. 2008).
In humans, we mainly draw upon case studies based on patients with RBD resulting from inflammatory or ischemic lesions incidentally involving the subcoeruleus region (the homologous structure to the SLD in humans) and its immediate surroundings, such as the dorsal pontomesencephalic area (Culebras and Moore 1989; Tippmann-Peikert et al. 2006), the pontine tegmentum (Mathis et al. 2007; Limousin et al. 2009; Xi and Luning 2009), and the paramedian caudal pons (Kimura et al. 2000).
In addition, diffusion-tensor imaging studies focusing on neuronal fiber integrity showed structural damages of pontomesencephalic structures in patients with idiopathic RBD (Unger et al. 2010; Scherfler et al. 2011). New-onset RBD was also reported in patients with lesions or structural changes in more rostral regions, such as the limbic system, the thalamus and the hypothalamus, apparently causing dysfunctions of the brainstem REM sleep areas (Provini et al. 2004; Boeve et al. 2007; Iranzo and Aparicio 2009; Unger et al. 2010).
Certainly, the conclusiveness of these studies is limited in particular as (1) none of these lesions is restricted to a particular anatomic structure, (2) they do not provide information about the neurochemical characteristics of the lesioned structures and projections, (3) it remains unclear whether all patients with those lesions develop RBD and whether all RBD patients have lesions in this particular region.
Nevertheless, those studies provide valuable insights especially into the neuroanatomical substrates of RBD, and with a rising awareness of this parasomnia as well as future high-resolution imaging techniques we may further enhance our understanding of RBD. Additionally, they set an example of the importance of cooperation and information exchange between basic and clinical researchers in translational research.
In contrast to the similarities in motor behavior, differences regarding REM sleep as such exist. All animal species with lesions or genetic modifications confined to the SLD showed loss of REM atonia, most of which without a decrease in REM sleep time. However, combined lesions or Vglut knockout of the SLD and its adjacent region (caudal laterodorsal tegmental nucleus, cLDT) resulted in an additional loss of atonia and a reduction of the amount of REM sleep and/or its stability, which is why the SLD-cLDT is considered a “REM sleep generator” and the SLD a “atonia generator” (Jouvet 1965; Webster and Jones 1988; Luppi et al. 2006; Lu et al. 2006; Sapin et al. 2009; Krenzer et al. 2011). Patients with idiopathic RBD typically do not show a significant reduction or fragmentation of REM sleep even despite severe movements (Iranzo and Aparicio 2009). Therefore, the SLD, rather than the SLD-cLDT seems affected in patients with RBD. Certainly, further studies are required on this particular issue. However, if the amount of REM sleep in patients with idiopathic RBD was not altered indeed, it would be more than interesting to determine why descending neurons are damaged in particular. Under the assumption that neurodegenerative processes proceed rostrocaudally along neuronal projections, ascending REM-modulating pathways originating from the glutamatergic extended SLD (including the caudal laterodorsal pontine tegmentum and targeting the cortex via projections to the parabrachial nucleus and the basal forebrain), might only be affected in later stages or to a smaller extent in humans.
In addition, the degeneration of dopaminergic neurons might not only play a role in manifesting Parkinson’s disease, but also in its precursor RBD, as indicated by a marmoset MPTP model (Verhave et al. 2011). The latter is consistent with the frequent co-morbidity of PD and RBD. However, this model cannot explain why some patients with Parkinson’s disease, especially the ones with a rather mild form, do not show any signs of RBD and why the RBD phenotype disappears in others prior to the onset of Parkinson’s disease (Lai and Siegel 2003; Iranzo and Aparicio 2009).
The ventromedial medulla (VMM)
Many studies have suggested that the ventromedial medulla or its subregions might relay SLD projections to motor neurons (Sastre et al. 1981; Chase and Morales 1990). Using anatomical and pharmacological techniques, the pontine neurons projecting to the VMM were further characterized as glutamatergic (Lai and Siegel 1988; Lai et al. 1999). This was supported by findings that glutamatergic release within the ventromedial medulla increases during REM sleep, that its electrical as well as pharmacological stimulation with glutamate and non-NMDA agonists resulted in a reduced muscle tone, whereas cytotoxic lesions lead to an increased muscle tone (Lai and Siegel 1988, 1991; Schenkel and Siegel 1989; Holmes and Jones 1994; Kodama et al. 1998; Hajnik et al. 2000).
The ventromedial medulla contains GABA and glycinergic neurons, which are active during REM sleep and project to spinal motoneurons (Holstege and Bongers 1991; Boissard et al. 2002; Morales et al. 2006). Consequently, glutamatergic projections originating from the SLD target those inhibitory premotor VMM neurons, which directly inhibit alpha motoneurons (Fort et al. 2009). On the other hand, the VMM also exhibits glutamatergic neurons, which modulate REM atonia by activation of inhibitory interneurons at the spinal level (Vetrivelan et al. 2009). Since different subregions and transmitter systems of the VMM were investigated in these studies, their results are not necessarily contradictive to each other.
In view of studies in humans, only one clinical case of a medullary lesion associated with new-onset RBD was found, and this patient also had pontine inflammations sufficient to explain the pathophysiology (Limousin et al. 2009). According to the Braak staging of the Parkinson’s disease-related pathology, a lesion in this region would correspond to the early stage II without clinical symptoms (Braak et al. 2004). Even though Braak did not focus on sleep, we presumably miss those cases in the clinical setting, indeed, as the VMM seems to modulate rather than actually generate REM atonia. As a result, patients with medullary dysfunctions might primarily show cardiovascular and other autonomic dysfunctions rather than RBD.
The spinal ventral horn
Like in other types of motor behavior, cranial efferents target alpha motor neurons at the spinal level.
With regard to the regulation of REM atonia, a number of monoaminergic, orexinergic and MCHergic neurons were discovered, which target the spinal cord either directly or indirectly via the ventrolateral periaqueductal grey (vlPAG), the SLD or the VMM (Lai et al. 2001; Mileykovskiy et al. 2002; Kodama et al. 2003; Willie et al. 2008; Hassani et al. 2009, 2010, Luppi et al. 2011; Peever 2011).
From a clinical point of view, a disruption of these pathways may explain the occurence of RBD due to treatment with antidepressants and cases with a co-morbidity of RBD and narcolepsy (Schenck and Mahowald 1992; Gagnon et al. 2006; Dauvilliers et al. 2007; Luppi et al. 2011; Ju et al. 2012).
Other descending projections originating from glutamatergic SLD REM on areas either target GABA/glycinergic spinal interneurons directly, indirectly via the glutamatergic VMM or project to inhibitory interneurons in the VMM, which in turn target alpha motor neurons (Lu et al. 2006; Luppi et al. 2006; Fort et al. 2009; Vetrivelan et al. 2009).
Ultimately, alpha motor neurons are inhibited by glycine and GABA. The glycinergic involvement in the regulation of REM atonia at a spinal level has long been established in animal models using a huge variety of methods (Chase et al. 1989; Soja et al. 1991; Kohlmeier et al. 1996; Taepavarapruk et al. 2008; Chase 2008; Brooks and Peever 2011).
The role of GABA and its corresponding receptors was further clarified recently, showing that metabotropic GABAB and ionotropic GABAA/glycine receptors together are necessary to mediate REM sleep atonia (Brooks and Peever 2012).
Today the first line therapeutic option for the treatment of RBD is clonazepam, a GABAA receptor agonist (Aurora et al. 2010). Based on the findings by Brooks and Peever, we do not only further understand the therapeutic mechanism of action, but might also find out whether a combination of clonazepam and a GABAB receptor agonist might be beneficial from a clinical point of view.
In conclusion, our current understanding of the pathomechanisms underlying REM sleep behavior disorder is founded on basic animal research, but could only be extended by means of a constant interaction between basic research and clinical findings. As the open questions raised in this paper indicate, this interaction will become even more important in the future in order to further elucidate the pathomechanisms of human RBD and, eventually, create an early symptomatic, if not a disease modifying, therapeutic window for neurodegenerative disorders.
References
American Academy of Sleep Medicine (2005) The international classification of sleep disorders: diagnostic and coding manual, Westchester Ill. American Academy of Sleep Medicine
Aurora RN, Zak RS, Maganti RK, Auerbach SH, Casey KR, Chowdhuri S, Karippot A, Ramar K, Kristo DA, Morgenthaler TI (2010) Best practice guide for the treatment of REM sleep behavior disorder (RBD). J Clin Sleep Med 6:85–95
Boeve BF (2010) REM sleep behavior disorder: updated review of the core features, the REM sleep behavior disorder–neurodegenerative disease association, evolving concepts, controversies, and future directions. Ann N Y Acad Sci 1184:15–54
Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, Benarroch EE, Ahlskog JE, Smith GE, Caselli RC, Tippman-Peikert M, Olson EJ, Lin SC, Young T, Wszolek Z, Schenck CH, Mahowald MW, Castillo PR, del Tredici K, Braak H (2007) Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 130:2770–2788
Boissard R, Gervasoni D, Schmidt MH, Barbagli B, Fort P, Luppi PH (2002) The rat ponto-medullary network responsible for paradoxical sleep onset and maintenance: a combined microinjection and functional neuroanatomical study. Eur J Neurosci 16:1959–1973
Braak H, Ghebremedhin E, Rub U, Bratzke H, del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318:121–134
Brooks PL, Peever JH (2011) Impaired GABA and glycine transmission triggers cardinal features of rapid eye movement sleep behavior disorder in mice. J Neurosci 31:7111–7121
Brooks PL, Peever JH (2012) Identification of the transmitter and receptor mechanisms responsible for REM sleep paralysis. J Neurosci 32:9785–9795
Chase MH (2008) Confirmation of the consensus that glycinergic postsynaptic inhibition is responsible for the atonia of REM sleep. Sleep 31:1487–1491
Chase MH, Morales FR (1990) The atonia and myoclonia of active (REM) sleep. Annu Rev Psychol 41:557–584
Chase MH, Soja PJ, Morales FR (1989) Evidence that glycine mediates the postsynaptic potentials that inhibit lumbar motoneurons during the atonia of active sleep. J Neurosci 9:743–751
Clement O, Sapin E, Berod A, Fort P, Luppi PH (2011) Evidence that neurons of the sublaterodorsal tegmental nucleus triggering paradoxical (REM) sleep are glutamatergic. Sleep 34:419–423
Culebras A, Moore JT (1989) Magnetic resonance findings in REM sleep behavior disorder. Neurology 39:1519–1523
Dauvilliers Y, Rompre S, Gagnon JF, Vendette M, Petit D, Montplaisir J (2007) REM sleep characteristics in narcolepsy and REM sleep behavior disorder. Sleep 30:844–849
Fort P, Bassetti CL, Luppi PH (2009) Alternating vigilance states: new insights regarding neuronal networks and mechanisms. Eur J Neurosci 29:1741–1753
Gagnon JF, Postuma RB, Montplaisir J (2006) Update on the pharmacology of REM sleep behavior disorder. Neurology 67:742–747
Hajnik T, Lai YY, Siegel JM (2000) Atonia-related regions in the rodent pons and medulla. J Neurophysiol 84:1942–1948
Hassani OK, Lee MG, Henny P, Jones BE (2009) Discharge profiles of identified GABAergic in comparison to cholinergic and putative glutamatergic basal forebrain neurons across the sleep-wake cycle. J Neurosci 29:11828–11840
Hassani OK, Henny P, Lee MG, Jones BE (2010) GABAergic neurons intermingled with orexin and MCH neurons in the lateral hypothalamus discharge maximally during sleep. Eur J Neurosci 32:448–457
Henley K, Morrison AR (1974) A re-evaluation of the effects of lesions of the pontine tegmentum and locus coeruleus on phenomena of paradoxical sleep in the cat. Acta Neurobiol Exp (Wars) 34:215–232
Holmes CJ, Jones BE (1994) Importance of cholinergic, GABAergic, serotonergic and other neurons in the medial medullary reticular formation for sleep–wake states studied by cytotoxic lesions in the cat. Neuroscience 62:1179–1200
Holstege JC, Bongers CM (1991) A glycinergic projection from the ventromedial lower brainstem to spinal motoneurons. An ultrastructural double labeling study in rat. Brain Res 566:308–315
Iranzo A, Aparicio J (2009) A lesson from anatomy: focal brain lesions causing REM sleep behavior disorder. Sleep Med 10:9–12
Jouvet M (1965) Paradoxical sleep—a study of its nature and mechanisms. Prog Brain Res 18:20–62
Ju YE, Larson-Prior L, Duntley S (2012) RBD and antidepressants. Sleep Med 13:211–212
Kimura K, Tachibana N, Kohyama J, Otsuka Y, Fukazawa S, Waki R (2000) A discrete pontine ischemic lesion could cause REM sleep behavior disorder. Neurology 55:894–895
Kodama Y, Iwase S, Mano T, Cui J, Kitazawa H, Okada H, Takeuchi S, Sobue G (1998) Attenuation of regional differentiation of sympathetic nerve activity during sleep in humans. J Auton Nerv Syst 74:126–133
Kodama T, Lai YY, Siegel JM (2003) Changes in inhibitory amino acid release linked to pontine-induced atonia: an in vivo microdialysis study. J Neurosci 23:1548–1554
Kohlmeier KA, Lopez-Rodriguez F, Liu RH, Morales FR, Chase MH (1996) State-dependent phenomena in cat masseter motoneurons. Brain Res 722:30–38
Krenzer M, Anaclet C, Vetrivelan R, Wang N, Vong L, Lowell BB, Fuller PM, Lu J (2011) Brainstem and spinal cord circuitry regulating REM sleep and muscle atonia. PLoS ONE 6:e24998
Lai YY, Siegel JM (1988) Medullary regions mediating atonia. J Neurosci 8:4790–4796
Lai YY, Siegel JM (1991) Pontomedullary glutamate receptors mediating locomotion and muscle tone suppression. J Neurosci 11:2931–2937
Lai YY, Siegel JM (2003) Physiological and anatomical link between Parkinson-like disease and REM sleep behavior disorder. Mol Neurobiol 27(2):137–152
Lai YY, Clements JR, Wu XY, Shalita T, Wu JP, Kuo JS, Siegel JM (1999) Brainstem projections to the ventromedial medulla in cat: retrograde transport horseradish peroxidase and immunohistochemical studies. J Comp Neurol 408:419–436
Lai YY, Kodama T, Siegel JM (2001) Changes in monoamine release in the ventral horn and hypoglossal nucleus linked to pontine inhibition of muscle tone: an in vivo microdialysis study. J Neurosci 21:7384–7391
Lai YY, Hsieh KC, Nguyen D, Peever J, Siegel JM (2008) Neurotoxic lesions at the ventral mesopontine junction change sleep time and muscle activity during sleep: an animal model of motor disorders in sleep. Neuroscience 154:431–443
Limousin N, Dehais C, Gout O, Heran F, Oudiette D, Arnulf I (2009) A brainstem inflammatory lesion causing REM sleep behavior disorder and sleepwalking (parasomnia overlap disorder). Sleep Med 10:1059–1062
Lu J, Sherman D, Devor M, Saper CB (2006) A putative flip-flop switch for control of REM sleep. Nature 441:589–594
Luppi PH, Gervasoni D, Verret L, Goutagny R, Peyron C, Salvert D, Leger L, Fort P (2006) Paradoxical (REM) sleep genesis: the switch from an aminergic–cholinergic to a GABAergic–glutamatergic hypothesis. J Physiol Paris 100:271–283
Luppi PH, Clement O, Sapin E, Gervasoni D, Peyron C, Leger L, Salvert D, Fort P (2011) The neuronal network responsible for paradoxical sleep and its dysfunctions causing narcolepsy and rapid eye movement (REM) behavior disorder. Sleep Med Rev 15:153–163
Mathis J, Hess CW, Bassetti C (2007) Isolated mediotegmental lesion causing narcolepsy and rapid eye movement sleep behaviour disorder: a case evidencing a common pathway in narcolepsy and rapid eye movement sleep behaviour disorder. J Neurol Neurosurg Psychiatry 78:427–429
Mileykovskiy BY, Kiyashchenko LI, Siegel JM (2002) Muscle tone facilitation and inhibition after orexin-a (hypocretin-1) microinjections into the medial medulla. J Neurophysiol 87:2480–2489
Morales FR, Sampogna S, Rampon C, Luppi PH, Chase MH (2006) Brainstem glycinergic neurons and their activation during active (rapid eye movement) sleep in the cat. Neuroscience 142:37–47
Peever J (2011) Control of motoneuron function and muscle tone during REM sleep, REM sleep behavior disorder and cataplexy/narcolepsy. Arch Ital Biol 149:454–466
Postuma RB, Lang AE, Gagnon JF, Pelletier A, Montplaisir JY (2012) How does parkinsonism start? Prodromal parkinsonism motor changes in idiopathic REM sleep behaviour disorder. Brain 135:1860–1870
Provini F, Vetrugno R, Pastorelli F, Lombardi C, Plazzi G, Marliani AF, Lugaresi E, Montagna P (2004) Status dissociatus after surgery for tegmental ponto-mesencephalic cavernoma: a state-dependent disorder of motor control during sleep. Mov Disord 19:719–723
Sakai K, Sastre JP, Salvert D, Touret M, Tohyama M, Jouvet M (1979) Tegmentoreticular projections with special reference to the muscular atonia during paradoxical sleep in the cat: an HRP study. Brain Res 176:233–254
Sapin E, Lapray D, Berod A, Goutagny R, Leger L, Ravassard P, Clement O, Hanriot L, Fort P, Luppi PH (2009) Localization of the brainstem GABAergic neurons controlling paradoxical (REM) sleep. PLoS One 4:e4272
Sastre JP, Sakai K, Jouvet M (1981) Are the gigantocellular tegmental field neurons responsible for paradoxical sleep? Brain Res 229:147–161
Schenck CH, Mahowald MW (1992) Motor dyscontrol in narcolepsy: rapid-eye-movement (REM) sleep without atonia and REM sleep behavior disorder. Ann Neurol 32:3–10
Schenck CH, Bundlie SR, Ettinger MG, Mahowald MW (1986) Chronic behavioral disorders of human REM sleep: a new category of parasomnia. Sleep 9:293–308
Schenkel E, Siegel JM (1989) REM sleep without atonia after lesions of the medial medulla. Neurosci Lett 98:159–165
Scherfler C, Frauscher B, Schocke M, Iranzo A, Gschliesser V, Seppi K, Santamaria J, Tolosa E, Hogl B, Poewe W (2011) White and gray matter abnormalities in idiopathic rapid eye movement sleep behavior disorder: a diffusion-tensor imaging and voxel-based morphometry study. Ann Neurol 69:400–407
Soja PJ, Lopez-Rodriguez F, Morales FR, Chase MH (1991) The postsynaptic inhibitory control of lumbar motoneurons during the atonia of active sleep: effect of strychnine on motoneuron properties. J Neurosci 11:2804–2811
Taepavarapruk N, Taepavarapruk P, John J, Lai YY, Siegel JM, Phillips AG, McErlane SA, Soja PJ (2008) State-dependent changes in glutamate, glycine, GABA, and dopamine levels in cat lumbar spinal cord. J Neurophysiol 100:598–608
Tippmann-Peikert M, Boeve BF, Keegan BM (2006) REM sleep behavior disorder initiated by acute brainstem multiple sclerosis. Neurology 66:1277–1279
Unger MM, Belke M, Menzler K, Heverhagen JT, Keil B, Stiasny-Kolster K, Rosenow F, Diederich NJ, Mayer G, Moller JC, Oertel WH, Knake S (2010) Diffusion tensor imaging in idiopathic REM sleep behavior disorder reveals microstructural changes in the brainstem, substantia nigra, olfactory region, and other brain regions. Sleep 33:767–773
Verhave PS, Jongsma MJ, van den Berg RM, Vis JC, Vanwersch RA, Smit AB, van Someren EJ, Philippens IH (2011) REM sleep behavior disorder in the marmoset MPTP model of early Parkinson disease. Sleep 34:1119–1125
Vetrivelan R, Fuller PM, Tong Q, Lu J (2009) Medullary circuitry regulating rapid eye movement sleep and motor atonia. J Neurosci 29:9361–9369
Webster HH, Jones BE (1988) Neurotoxic lesions of the dorsolateral pontomesencephalic tegmentum-cholinergic cell area in the cat. II. Effects upon sleep-waking states. Brain Res 458:285–302
Willie JT, Sinton CM, Maratos-Flier E, Yanagisawa M (2008) Abnormal response of melanin-concentrating hormone deficient mice to fasting: hyperactivity and rapid eye movement sleep suppression. Neuroscience 156:819–829
Xi Z, Luning W (2009) REM sleep behavior disorder in a patient with pontine stroke. Sleep Med 10:143–146
Conflict of interest
The authors declare that they have no conflict of interest with regard to this paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Krenzer, M., Lu, J., Mayer, G. et al. From bench to bed: putative animal models of REM sleep behavior disorder (RBD). J Neural Transm 120, 683–688 (2013). https://doi.org/10.1007/s00702-012-0965-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-012-0965-x