
Abstract
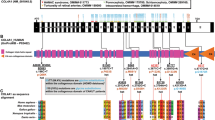
Uromodulin (UMOD) mutations were described in patients with medullary cystic kidney disease (MCKD2), familial juvenile hyperuricemic nephropathy (FJHN), and glomerulocystic kidney disease (GCKD). UMOD transcription is activated by the transcription factor HNF1B. Mutations in HNF1B cause a phenotype similar to FJHN/GCKD but also congenital anomalies of the kidney and the urinary tract (CAKUT). Moreover, we recently detected UMOD mutations in two patients with CAKUT. As HNF1B and UMOD act in the same pathway and cause similar phenotypes, we here examined whether UMOD mutations would be found in patients with CAKUT. Mutation analysis of UMOD was performed in 96 individuals with CAKUT by direct sequencing of exons 4 and 5 and by heteroduplex analysis following CEL I digestion assay of exons 3 and 6–12. Mean patient age was 11.4 years, and in 36.4% of patients, family history was positive for CAKUT. In the CEL I assay, 12 aberrant bands were detected in 103 of 960 polymerase chain reaction (PCR) products and were sequenced. Six previously known and seven new single nucleotide polymorphisms (SNPs) were detected. As no UMOD mutations were identified in these 96 patients with CAKUT, UMOD mutations do not seem to be a significant cause of CAKUT in this cohort.
Similar content being viewed by others


References
Tamm I, Horsfall FL Jr (1950) Characterization and separation of an inhibitor of viral hemagglutination present in urine. Proc Soc Exp Biol Med 74:106–108
Hart TC, Gorry MC, Hart PS, Woodard AS, Shihabi Z, Sandhu J, Shirts B, Xu L, Zhu H, Barmada MM, Bleyer AJ (2002) Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet 39:882–892
Dahan K, Devuyst O, Smaers M, Vertommen D, Loute G, Poux JM, Viron B, Jacquot C, Gagnadoux MF, Chauveau D, Buchler M, Cochat P, Cosyns JP, Mougenot B, Rider MH, Antignac C, Verellen-Dumoulin C, Pirson Y (2003) A cluster of mutations in the UMOD gene causes familial juvenile hyperuricemic nephropathy with abnormal expression of Uromodulin. J Am Soc Nephrol 14:2883–2893
Rampoldi L, Caridi G, Santon D, Boaretto F, Bernascone I, Lamorte G, Tardanico R, Dagnino M, Colussi G, Scolari F, Ghiggeri GM, Amoroso A, Casari G (2003) Allelism of MCKD, FJHN, and GCKD caused by impairment of uromodulin export dynamics. Hum Mol Genet 12:3369–3384
Wolf MT, Mucha BE, Attanasio M, Zalewski I, Karle SM, Neumann HP, Rahman N, Bader B, Baldamus CA, Otto E, Witzgall R, Fuchshuber A, Hildebrandt F (2003) Mutations of the Uromodulin gene in MCKD type 2 patients cluster in exon 4 which encodes three EGF-like domains. Kidney Int 64:1580–1587
Turner JJ, Stacey JM, Harding B, Kotanko P, Lhotta K, Puig JG, Roberts I, Torres RJ, Thakker RV (2003) Uromodulin mutations cause familial juvenile hyperuricemic nephropathy. J Clin Endocrinol Metab 88:464–470
Nakai S, Sugitani Y, Sato H, Ito S, Miura Y, Ogawa M, Nishi M, Jishage K, Minowa O, Noda T (2003) Crucial roles of Brn1 in distal tubule formation and function in mouse kidney. Development 130:4751–4759
Wolf MT, Beck BB, Zaucke F, Kunze A, Misselwitz J, Ruley J, Ronda T, Fischer A, Eifinger F, Licht C, Otto E, Hoppe B, Hildebrandt F (2007) The Uromodulin C744G mutation causes MCKD2 and FJHN in children and adults and may be due to a possible founder effect. Kidney Int 71:574–581
Gresh L, Fischer E, Reimann A, Tanguy M, Garbay S, Shao X, Hiesberger T, Fiette L, Igarashi P, Yaniv M, Pontoglio M (2004) A transcriptional network in polycystic kidney disease. EMBO J 23:1657–1668
Decramer S, Parant O, Beaufils S, Clauin S, Guillou C, Kessler S, Aziza J, Bandin F, Schanstra JP, Bellanne-Chantelot C (2007) Anomalies of the TCF2 gene are the main cause of fetal bilateral hyperechogenic kidneys. J Am Soc Nephrol 18:923–933
Salomon R, Moriniere V, Weber S, Wuehl E, Schaefer F, Antignac C (2004) PAX2, EYA1 and HNF1B mutations renal hypo-dysplasia, abstract. J Am Soc Nephrol 15:662A
Bellanne-Chantelot C, Chauveau D, Gautier JF, Dubois-Laforgue D, Clauin S, Beaufils S, Wilhelm JM, Boitard C, Noel LH, Velho G, Timsit J (2004) Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann Intern Med 140:510–517
Kaplan BS, Gordon I, Pincott J, Barratt TM (1989) Familial hypoplastic glomerulocystic kidney disease: a definite entity with dominant inheritance. Am J Med Genet 34:569–573
Bingham C, Ellard S, van’t Hoff WG, Simmonds HA, Marinaki AM, Badman MK, Winocour PH, Stride A, Lockwood CR, Nicholls AJ, Owen KR, Spyer G, Pearson ER, Hattersley AT (2003) Atypical familial juvenile hyperuricemic nephropathy associated with a hepatocyte nuclear factor-1beta gene mutation. Kidney Int 63:1645–1651
Wolf MTF, Saunier S, O’Toole JF, Wanner N, Groshong T, Attanasio M, Salomon R, Stallmach T, Sayer JA, Waldherr R, Griebel M, Oh J, Neuhaus TJ, Josefiak U, Antignac C, Otto EA, Hildebrandt F (2007) Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephronophthisis. Kidney Int 72:1520–1526
Oleykowski CA, Bronson Mullins CR, Godwin AK, Yeung AT (1998) Mutation detection using a novel plant endonuclease. Nucleic Acids Res 26:4597–4602
Otto EA, Helou J, Allen SJ, O’Toole JF, Attanasio M, Zhou W, Wolf MTF, Hildebrandt F (2007) Mutation analysis in nephronophthisis using a combined approach of homozygosity mapping, CEL I endonuclease cleavage, and direct sequencing. Hum Mutat 29:418–426
NCBI, National Center for Biotechnology Information, Entrez Single Nucleotide Polymorphism Database. Available at https://doi.org/www.ncbi.nlm.nih.gov/sites/entrez?db=snp. Accessed on 13 April 2008
Schedl A (2007) Renal abnormalities and their developmental origin. Nat Rev Genet 8:791–802
Weber S, Taylor JC, Winyard P, Baker KF, Sullivan-Brown J, Schild R, Knüppel T, Zurowska AM, Caldas-Alfonso A, Litwin M, Emre S, Ghiggeri GM, Bakkaloglu A, Mehls O, Antignac C, Network E, Schaefer F, Burdine RD (2008) SIX2 and BMP4 mutations associate with anomalous kidney development. J Am Soc Nephrol 19:891–903
Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, Jankauskiené A, Mir S, Montini G, Peco-Antic A, Wühl E, Zurowska AM, Mehls O, Antignac C, Schaefer F, Salomon R (2006) Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol 17:2864–2870
Lu W, van Eerde AM, Fan X, Quintero-Rivera F, Kulkarni S, Ferguson H, Kim HG, Fan Y, Xi Q, Li QG, Sanlaville D, Andrews W, Sundaresan V, Bi W, Yan J, Giltay JC, Wijmenga C, de Jong TP, Feather SA, Woolf AS, Rao Y, Lupski JR, Eccles MR, Quade BJ, Gusella JF, Morton CC, Maas RL (2007) Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am J Hum Genet 80:616–632
Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, Kumar S, Neuhaus TJ, Kemper MJ, Raymond RM Jr, Brophy PD, Berkman J, Gattas M, Hyland V, Ruf EM, Schwartz C, Chang EH, Smith RJ, Stratakis CA, Weil D, Petit C, Hildebrandt F (2004) SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci U S A 101:8090–8095
Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, Dobyns WB, Eccles MR (1995) Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet 9:358–364
Hoskins BE, Cramer CH, Silvius D, Zou D, Raymond RM, Orten DJ, Kimberling WJ, Smith RJ, Weil D, Petit C, Otto EA, Xu PX, Hildebrandt F (2007) Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet 80:800–804
Pope JC 4th, Brock JW 3rd, Adams MC, Stephens FD, Ichikawa I (1999) How they begin and how they end: classic and new theories for the development and deterioration of congenital anomalies of the kidney and urinary tract, CAKUT. J Am Soc Nephrol 10:2018–2028
Acknowledgements
We thank all individuals who participated in this study. The technical assistance of Steffi Schneider is gratefully acknowledged. FH is the Frederick G.L. Huetwell professor and a Doris Duke Distinguished Clinical Scientist. He is supported by grants from the NIH (DK068306, DK064614 and DK069274). MTFW was supported by grants from the Koeln Fortune Program Faculty of Medicine, University of Cologne (184/2004), the German Kidney Fund (Deutsche Nierenstiftung), and the German Research Foundation (DFG WO 1229/2-1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wolf, M.T.F., Hoskins, B.E., Beck, B.B. et al. Mutation analysis of the Uromodulin gene in 96 individuals with urinary tract anomalies (CAKUT). Pediatr Nephrol 24, 55–60 (2009). https://doi.org/10.1007/s00467-008-1016-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-008-1016-6