
Abstract
SH3 and cysteine-rich protein 3 (STAC3), a small adapter protein originally identified as a core component of excitation–contraction coupling machinery, regulates the voltage-induced Ca2+ release in skeletal muscle. However, the possibility of additional, as yet unknown, non-muscle effects of STAC3 cannot be ruled out. Herein, we provide the evidence for the expression and functional involvement of STAC3 in spermatogenesis. STAC3 expression was localized in the testicular interstitium of rodent and human testes. By using the cytotoxic drug ethylene dimethane sulfonate (EDS), STAC3 expression was observed to be decreased sharply in rat testis after selective withdrawal of Leydig cells (LCs), and reappeared immediately after LCs repopulation, indicating that testicular expression of STAC3 mainly stems from LCs. From a functional standpoint, in vivo lentiviral vector–mediated suppression of STAC3 resulted in a significant decrease in testosterone production, and thereafter caused impairment of male fertility by inducing oligozoospermia and asthenospermia. The indispensible involvement of STAC3 in testicular steroidogenesis was validated using the in vivo knockdown model with isolated primary LCs as well as in vitro experiments with primary LCs. By generating the TM3Stac3−/− cells, we further revealed that STAC3 depletion attenuated mitochondrial membrane potential and StAR processing in db-cAMP-stimulated LCs. Thus, the inhibitory effect of STAC3 deficiency on testicular steroidogenesis may be ascribed to a disturbed mitochondrial homeostasis. Collectively, the present results strongly suggest that STAC3 may function as a novel regulator linking mitochondrial homeostasis and testicular steroidogenesis in LCs. Our data underscore an unexpected reproductive facet of this muscle-derived factor.
Similar content being viewed by others


Introduction
Interstitial Leydig cells (LCs) are the main source for biosynthesized testosterone, and proper production of testosterone is essential for germ cell survival and differentiation (Goldberg, 1984; Holdcraft and Braun, 2004). Upon stimulation with luteinizing hormone (LH), cyclic adenosine monophosphate (cAMP) accumulates rapidly in LCs, where cAMP acts as an intracellular second messenger to regulate testosterone synthesis either acutely at the level of cholesterol transport or chronically at the level of gene transcription (Zirkin and Papadopoulos, 2018). In response to intracellular pulse by cAMP, steroidogenic acute regulatory protein (StAR) is expressed as a 37-kDa precursor outside mitochondrion, and is then imported into the mitochondrial matrix where it is processed into a mature 30-kDa form. StAR facilitates the transfer of cholesterol into mitochondrial matrix to fuel the testosterone biosynthesis. Therefore, StAR serves as the first and rate-limiting step of complicated testicular steroidogenesis (Stocco and Clark, 1996). Accumulated data suggest that the phosphorylation of StAR is critical in order to render the protein fully active in its capacity to support cholesterol transfer (Arakane, et al., 1997). Moreover, during aerobic respiration, ATP is generated and electrons are transported through enzyme complexes (nicotinamide adenine dinucleotide hydroxide dehydrogenase, succinate dehydrogenase, cytochrome bc1 and cytochrome oxidase) that are located at the inner mitochondrial membrane. This electron transportation yields free energy that generates a proton gradient. The proton gradient then promotes a pH differential (ΔpH) and a mitochondrial membrane potential (ΔΨm), both of which provide the proton motive force driving ATP synthesis (Allen, et al., 2006). Collective findings suggest that proper ATP production generated by mitochondria is also crucial for steroid biosynthesis (Midzak, et al., 2011).
SH3 and cysteine-rich protein 3 (STAC3), a small adapter protein, was originally identified as a core component of excitation–contraction (EC) coupling machinery in skeletal muscle (Horstick, et al., 2013). It mediates voltage-induced Ca2+ release and contractility. Homozygous deletion of the Stac3 gene results in complete paralysis and perinatal lethality with a range of musculoskeletal defects in mice (Nelson, et al., 2013). Likewise, mutations in the human STAC3 gene lead to myopathy (Horstick, et al., 2013). To be noted, the biological effects of STAC3 known to date are largely performed in skeletal muscles. However, additional as yet unknown non-muscle actions of STAC3 could not be ruled out. In this regard, using panel-based nuclear and mitochondrial next-generation sequencing, aberrant expression of STAC3 has been associated with mitochondrial diseases in pediatric patients (Schoonen, et al., 2019). Novel expression of STAC has been reported in central nerve system (Polster, et al., 2018). Additionally, using Xenopus oocytes, Wu F et al. observe that Stac3 enhances expression of human CaV1.1, and helps to reveal gating pore currents in HypoPP mutant channels (Wu, et al., 2018). Nevertheless, the functional details of STAC3 in such non-muscle systems remain largely unexplored.
It is now evident that cellular Ca2+ homeostasis plays a central role in the modulation of diverse cellular functions in both germ cells and somatic cells within mammalian testis (Lee, et al., 2011). The identification of STAC3 as a potent regulator of Ca2+ homeostasis prompted us to evaluate whether this signaling is functionally expressed in testis. Our systematic analysis demonstrates that STAC3 may act as a novel factor linking mitochondrial homeostasis and testicular steroidogenesis, thus underscoring an unexpected reproductive facet of this novel adapter protein.
Materials and methods
Data mining
To explore the clinical relevance of STAC3 expression, we performed data mining on the data sheet GSE45885, analysis on testicular biopsy specimens from normozoospermia (n = 4) or from non-obstructive azoospermia (n = 27) using Affymetrix Human Gene 1.0 ST Array (Malcher, et al., 2013). To computerize the expression level, raw files were imported into the GEO2R online software provided by the GEO database (Qian, et al., 2019).
Human samples
Upon receipt of informed consent, normal human testicular specimens were collected from normozoospermic patients who underwent testicular biopsies during genital surgery procedures for varicocele or epididymal cysts (n = 12). Normal spermatogenesis was validated by histological examination and semen analysis (Tian, et al., 2014). Testicular tissues were immediately fixed in 4% paraformaldehyde (Sigma-Aldrich, Shanghai, China) and were embedded in paraffin. Partial samples were immediately snap-frozen in liquid nitrogen, stored at -80 °C for later biochemical analysis. The procedures involved in human study, strictly conformed to the 1964 Helsinki Declaration, were approved by the Human Research Committee of Children’s Hospital of Shanxi and Women Health Center of Shanxi (Approval #: CHSX-2014-00164b).
Animal model
Experimental procedures for the humane treatment and well-being of the laboratory animals, in compliance with the Guidelines for the Care and Use of Experimental Animals from NIH, were approved by the Institutional Animal Care and Use Committee of Children’s Hospital of Shanxi and Women Health Center of Shanxi (Approval #: CHSX-2014-00164a). All surgeries in animals were performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
C57BL/6 J male mice and 3-month-old male SD rats were obtained from the animal facility of our hospital. Animals were housed under environmentally controlled conditions (12 h light–dark cycle and 20–25 °C) with food and water provided ad libitum. They were allowed to acclimatize for at least 7 d before experiment.
Elimination of rat LCs was achieved by systemic administration of a single dose of ethylene dimethane sulfonate (EDS) (75 mg/kg i.p.) or vehicle control (dimethyl sulfoxide, water; 1.5:3.5, vol/vol), as described (He, et al., 2016). Testes and blood samples were harvested at 7, 14 and 28 d following EDS injection.
In vivo knockdown of Stac3 was carried out as described elsewhere (Kim, et al., 2010). After anaesthetization, testes were exposed and injected with 20 μl pLKO.1-CMV-tGFP-Stac3 (7 ng/μl) or empty vector (Sigma-Aldrich) using a fiber optics probe (diameter, 1.65 mm), under a Leica EZ4 dissecting microscope (Leica, Beijing, China). The testes were then returned to scrotum, incision was sutured, and mice were allowed to recover. Two days after microinjection, some mice were i.p. injected with testosterone propionate (TP, 3 mg/kg, Sigma-Aldrich) on a daily basis for consecutive 68 d. Testes and blood samples were harvested at 70 d after the first microinjection.
Cell treatment
Primary LCs were isolated and purified according to a reported protocol (Riccetti, et al., 2017). Briefly, testes from adult mice were decapsulated, mechanically dissociated, and were subjected to enzymatic digestion by 20 mg/ml collagenase (Sigma-Aldrich) under a constant shacking at 37 °C for 20 min. The resultant cell suspension was then filtered using a 100 μm Nylon mesh (Thermo Fisher Scientific, Shanghai, China). The collected LCs were subjected to a series of purification steps involving 0–100% v/v Percoll linear density gradient (Sigma-Aldrich). The purity of LCs was verified using 3β-hydroxysteroid dehydrogenase (HSD3B) immunostaining (Riccetti, et al., 2017), along with qPCR analysis using primer sets specific to germ cells, Sertoli cells, and LCs marker genes (Sun, et al., 2013). The TM3 mouse Leydig tumor cell line, obtained from American Type Culture Collection (ATCC, Manassas, VA, USA), was routinely maintained in a 1:1 mixture of Ham's F12 medium and Dulbecco's modified eagle's medium supplemented with 2.5 mM L-Glutamine, 0.5 mM Sodium Pyruvate, 1.2 g/L sodium bicarbonate and 15 mM HEPES, 92.5% horse serum, 5% FBS (Thermo Fisher Scientific), as instructed by the manufacturer. The Stac3-null TM3 cells were generated by transfection of pLKO.1-CMV-tGFP-Stac3, followed by selection with 0.5 μg/ml G418 (Thermo Fisher Scientific). To study the hormone-stimulated steroidogenesis, LCs or TM3 cells were incubated for 12 h with 100 ng/ml luteinizing hormone (LH)/human chorionic gonadotropin (hCG), 0.5 mmol/l db-cAMP, 5 μmol/l 22R-hydroxycholesterol (22-ROH), or 5 μmol/l pregnenolone respectively (all reagents were obtained from Sigma-Aldrich), followed by hormone assay.
Hormone assay
Serum testosterone levels in mice and concentrations of testosterone in the cell culture media were measured using a testosterone enzyme-linked immunosorbent assay (ELISA) kit (DRG, Marburg, Germany), as per the manufacturer's instructions. Final absorbance values were determined in triplicate at 450 nm on a microplate reader (Bio-Rad, Shanghai, China). The sensitivity of testosterone assay is 0.083 ng/mL. The intra-assay precision of these results is between 2.1 and 3.6%, and the inter-assay precision of is between 4.1 and 5.4%.
RT-qPCR
Total RNA was extracted using E.Z.N.A.® HP Total RNA Kit (Omega Bio-tek, Guangzhou, China). Following routine DNase digestion for RNA purification, 800 ng of RNA was subjected to the synthesis of the first strand cDNA using the iScript™ cDNA Synthesis Kit (Bio-Rad). Subsequent qPCR analysis using ~ 10 ng cDNA/reaction was carried out according to the Promega protocol. The relative expression levels of target genes were calculated using the 2−∆∆Ct method (Dong, et al., 2016), with amplification of 18S as the internal control. The primers used were: mouse Stac3 (NM_177707.4), 5′-CCCACTCTACAGCGACCAAC-3′ and 5′-GACTCTGGCTCCTCCTCCAT-3′; rat stac3 (NM_001130558.1), 5′-ACTTCAGCTCTACCACCACG-3′ and 5′-CTCCATCTCGGTTGTCTCTGG-3′; human STAC3 (NM_145064.3), 5′-TTTCAGCCCTGACACCAAGG-3′ and 5′-ACTTTGCCGAGTCTCTGCTG-3′; human 18S (M10098.1, also applied to mouse and rat 18 s), 5′-AATCAGGGTTCGATTCCGGA-3′ and 5′-GTGGACTCATTCCAATTACAG-3′. Primers used for Pem, Star, Hsd3b and Cyp11a1 have been reported elsewhere (Wang, et al., 2006, 2018).
Immunoblotting/Western blotting
Protein samples were prepared using a Chemicon® kit (Merck, Hong Kong, China). ~ 30 μg of protein samples were separated by SDS/PAGE, and were transferred to a PVDF membrane (Thermo Fisher Scientific). Membranes were then incubated at 4 °C overnight with different primary antibodies (Table 1). Positive signals were finally developed using the Immun-Star HRP chemiluminescence detection kit (Bio-Rad). Immunoblots hybridized with a preabsorbed serum (Abcam, Shanghai, China) served as negative controls.
Histological examination and measurement of testicular apoptosis
5-μm thick testicular sections were deparaffinized, rehydrated, and stained with hematoxylin–eosin for histological examination. To reveal the localization of STAC3 protein in situ, 5-μm thick testicular sections were deparaffinized, rehydrated, and subjected to antigen retrieval (20 min at 95 °C in 10 nM citrate buffer, pH 6.0). The sections were immersed in 0.5% v/v H2O2 /methanol for 20 min to block endogenous peroxidase activity. After PBS washes, sections were incubated at 4 °C overnight with STAC3 antibody. Subsequent hybridization with second-antibody and avidin–biotin complex (ABC) was performed with the aid of the VECTASTAIN ELITE® ABC Kit (Vector Lab, Burlingame, CA, USA). Peroxidases were finally detected with 0.7 mg/ml 3–3′-diaminobenzidine tetrahydrochloride (Sigma-Aldrich) in 1.6 mg/ml urea hydrogen peroxide.
Double-immunofluorescent cytochemistry was performed as described (Li, et al., 2014). TM3 cells were labeled with 500 nM MitoTracker Red CMXRos (Thermo Fisher Scientific) in fresh culture media at 37 °C for 30 min, and were then fixed with 4% paraformaldehyde for 15 min, followed by incubation at room temperature for 30 min with blocking solution (10% donkey serum, 0.5% bovine serum albumin and 0.1% triton X-100 in PBS). Cells were then incubated at 4 °C overnight with different antibodies (Table 1), followed by incubation at room temperature with fluorescein isothiocyanate (FITC)-labeled and rhodamine-labeled second antibodies. Cells were finally incubated at room temperature with 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) for 10 min. Double-immunofluorescent signals were observed and recorded under an Axio Imager M1 inverted microscope (Zeiss, Shanghai, China).
Testicular apoptosis was assessed using an apoptosis enzyme-linked immunosorbent assay (ELISA) kit (Roche, Shanghai, China), following the manufacturer’s instructions.
Analysis of ΔΨm with TMRE
Mitochondrial ΔΨm in stimulated LCs was evaluated by measuring uptake and accumulation of the potentiometric tetramethylrhodamine ethyl ester dye (TMRE, Sigma-Aldrich) (Allen, et al., 2006). TM3 cells were cultured to a maximum of 75% confluency in a 96-well florescence assay plate (Thermo Fisher Scientific). After incubation with 0.5 mM db-cAMP for 6 h, cells were treated at 37 °C in 200 μl of a 50-nM solution of TMRE in serum-free medium for 20 min. Final TMRE fluorescence was measured using an excitation of 550 nm (540/25 nm filter) and an emission at 590 nm (590/20 nm filter) under the Axio Imager M1 inverted microscope (Zeiss).
Northern blot
A biotin-labeled RNA probe was produced by in vitro transcription reactions using Ambion's MAXIscript™ Kit, following the manufacturer’s instructions. RNA samples were prepared as described above. About 30 μg of RNA samples were separated by denaturing agarose gel electrophoresis, and were then transferred onto a Blotting-Nylon 66 membrane (Sigma-Aldrich). Subsequent Northern blot analysis was carried out with the aid of the NorthernMax™ and BrightStar™ BioDetect™ Nonisotopic Detection system (Thermo Fisher Scientific). Final visualization of positive signals was achieved through phosphorimaging (Typhoon, Mundelein, IL, USA).
Statistical analysis
Data are presented as mean ± S.E.M. Statistical significance was assessed by performing Student's t-test or one way analysis of variance (ANOVA) as appropriate, using a P-value of 0.05. Statistical analyses were performed with SPSS13.0 software package (SPSS Inc., Chicago, IL, USA).
Results
Testicular STAC3 is exclusively expressed by LCs
From the transcriptomic profiles of 4 normozoospermia and 27 non-obstructive azoospermia deposited in the GEO database (GSE45885), the expression levels of the STAC3 transcripts, were observed to be significantly downregulated in non-obstructive azoospermia compared to those in normozoospermia (P < 0.0001, Fig. 1a). This finding suggests that the expression of STAC3 may be associated with spermatogenesis. As an initial step to understand the function of STAC3, we established the expression pattern of STAC3 in testis. RT-PCR assay revealed a distinct expression of Stac3/STAC3 in rodent and human testes (Fig. 1b). Moreover, immunoblotting analysis demonstrated a single band of STAC3 protein in the whole blot, whereas blots treated with a preabsorbed primary antibody demonstrated no bands, confirming the assay specificity (Fig. 1c). Subsequent immunohistochemistry showed that testicular STAC3 is exclusively expressed by LCs (Fig. 1d, d’ and d’’).

Expression of STAC3 in rodent and human testes. (a) Curve of analytical results showing the expression levels of STAC3 transcripts in testicular biopsies from 4 normozoospermia and 27 nonobstructive azoospermia, as reported by GEO Database (GSE45885). (b) Representative RT-PCR assay of the expression levels of Stac3/STAC3 mRNA in rodent and human testes was presented. Amplification of 18S mRNA served as the internal control. The PCR assay without reverse transcription was also accompanied as negative control. (c) Immunoblotting analysis demonstrated a single band of STAC3 protein in the testicular lysates from adult mouse, rat and human, whereas preabsorption with blocking peptides totally abolished the positive immunostaining. (d-d’’) Sections of adult rodent and human testes immunostained with a rabbit anti-STAC3 polyclonal antibody and counterstained with hematoxylin. Clear immunostaining is observed in the testicular interstitium. d, mouse testis; d’, rat testis; d’’, human testis. LCs, Leydig cells. Inserted panel, immunostaining with blocking peptides totally abolished the positive staining. Bar = 50 μm
Verification of the STAC3 localization in rodent LCs
Because STAC3 expression in mammalian testes appeared to be relatively conserved, we used EDS-treated rat testis to further verify the localization of STAC3 in LCs. Elimination of LCs by EDS was confirmed by histological examination (Fig. 2a and a’) and by measurement of circulating testosterone concentrations (Fig. 2b). In accordance with the previous reports (He, et al., 2016; Tena-Sempere, et al., 2002), testosterone concentrations in EDS-treated rats dropped to nearly undetectable levels at d 7 and 14, and began to restore at d 21 following EDS administration. Intriguingly, testicular expression of STAC3 was significantly decreased at d 7 and 14 after EDS injection, with the lowest values being observed at d 14. By contrast, expression of STAC3 restored substantially at 21 following EDS injection, well consistent with the hormonal changes (Fig. 2c and d). Thus, testicular expression of STAC3 is exclusively enriched in LCs.

Verification of the STAC3 localization in LCs using a cytotoxic drug ethylene dimethane sulfonate (EDS). (a-a’) Representative H&E-stained transverse testis sections showing morphology at a, 0; a', 7 d after administration of EDS. Bar = 100 μm (b) Serum testosterone level (ng/ml) in EDS-treated rats was evaluated as described in Materials and Methods (*P < 0.05 and **P < 0.01). (c) The expression levels of Stac3 mRNA in rat testis at different time points following EDS treatment were assessed by RT-qPCR. Relative expression levels were determined in which the level of a target mRNA was normalized against the mRNA level in mock control, which was arbitrarily set at 1. Data were presented as the mean ± S.E.M. of at least 3 determinations. (d) Levels of STAC3 protein in rat testis at different time points following EDS treatment were assessed by immunoblotting. TUBULIN was used as the loading control
Effects of in vivo inhibition of STAC3 expression on testicular steroidogenesis and spermatogenesis
It is becoming increasingly evident that microinjection of lentiviral plasmids could achieve an efficient in vivo gene transfer in LCs (either ectopic overexpression or targeted knockdown). This newly developed method therefore is a valuable tool to address the function of specific genes in testicular steroidogenesis (Kim, et al., 2010; Park, et al., 2013). To study the potential role of STAC3 in LCs at the in vivo level, we microinjected pLKO.1-CMV-tGFP-Stac3 or empty vector into the seminiferous tubules of adult C57BL/6 J mice. We then studied the expression of Stac3, testicular histology, fertility potential, testicular apoptosis and plasma testosterone concentrations in different experimental groups (Fig. 3a). Using a GFP reporter allele to evaluate targeting efficiency and specificity, we observed that the lentivirus-mediated bioluminescence was strongly enriched in the testicular interstitium at 35 d and was moderately expressed in the testicular interstitium at 70 d following microinjection, compared to the negative expression of bioluminescence in untreated testis (Fig. 3b, b’ and b’’). In accordance with these findings, at 70 d following microinjection, Stac3/STAC3 expression was significantly reduced in the testes injected with pLKO.1-CMV-tGFP-Stac3 compared with mock testes or those injected with empty vector, as revealed by RT-qPCR (Fig. 3c) and immunohistochemistry (Fig. 3d, d’, d’’ and d’’’). By contrast, supplement with testosterone propionate by s.c. injection on a daily basis failed to rescued the Stac3/STAC3 expression levels in the lentivirus-treated testes, suggesting that testicular expression of STAC3 is not regulated by androgen signaling. At 70 d following microinjection, mock testes exhibited normal histological features and high Johnsen’s scores, while Stac3 shRNA-treated testes demonstrated degenerative changes including germ cells desquamation, decrease of germinal epithelium height, and lower Johnsen’s scores (Fig. 3e, e’, e’’ and e’’’, Table 2). Consequently, inhibition of STAC3 expression caused impaired male fertility by inducing oligozoospermia and asthenospermia (Table 2). The stimulatory effect of Stac3 shRNA on testicular apoptosis, along with its inhibitory effect on androgen production, was also detected by ELISA at 70 d following microinjection (Fig. 3f and g). More importantly, the deleterious effects of Stac3 shRNA on testicular histoarchitecture, epididymal parameters, fertility potential, and testosterone production, were all successfully ameliorated by co-treatment with testosterone propionate (Fig. 3e, f, g and Table 2). For the selected genes expressed in testis, we observed that the expression levels of Pem (a well-defined androgen target gene) (Wang, et al., 2006) and Star (the first rate-limiting enzyme in testosterone synthesis) (Wang, et al., 2018) were reduced upon STAC3 ablation, whereas transcriptional levels of other rate-limiting enzymes (namely Hsd3b and Cyp11a1) were comparable in Ctrl sh and Stac3 sh-treated testes (Fig. 3h). These findings collectively suggest that STAC3 may regulate spermatogenesis by targeting the testicular steroidogenesis.

Effects of in vivo inhibition of STAC3 on testicular morphology, germ cell apoptosis, androgen production and male fertility. (a) Schematic representation of the experimental procedures used in the in vivo lentiviral vector–mediated shRNA treatment. (b-b’’) Enrichment of lentiviral shRNA in the testicular interstitium following microinjection was demonstrated by observing frozen sections under a fluorescence microscope. b, mock; b’, 35 d after microinjection; b’’, 70 d after microinjection. (c) Inhibition of Stac3 in testis by in vivo shRNA treatment was confirmed by RT-qPCR at 70 d after microinjection. Data were presented as the mean ± S.E.M. of at least 3 determinations (n = 10, *P < 0.05 and **P < 0.01). (d-d’’’) Immunostaining of STAC3 protein in testicular sections was carried out at different time points following microinjection. Bar = 25 μm (e-e’’’) Representative H&E-stained transverse testis sections showing morphological changes at different time points following microinjection. e, mock; e’, Ctrl sh; e’’, Stac3 sh; e’’’, Stac3 sh + testosterone propionate (TP). Bar = 50 μm (f) Effects of in vivo shRNA treatment on germ cell apoptosis at 70 d after microinjection were assessed using an ELISA kit (n = 10, *P < 0.05 and **P < 0.01). (g) Plasma testosterone levels (nmol/l) in mice from different groups at 70 d after microinjection were assessed using an ELISA method (n = 10, *P < 0.05 and **P < 0.01). (h) Effects of inhibition of Stac3 in testis by in vivo shRNA treatment on different target transcripts were revealed using RT-qPCR at 70 d after microinjection. Data were presented as the mean ± S.E.M. of at least 3 determinations (n = 5, *P < 0.05)
Validation of indispensible role of STAC3 in testicular steroidogenesis using the in vivo knockdown model with isolated primary LCs as well as in vitro experiments with TM3 and primary LCs
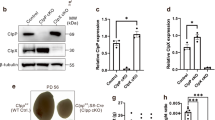
To validate the in vivo data, we isolated the primary LCs from Ctrl shRNA or Stac3 sh-treated testes (Supplementary Fig. 1, Fig. 4a). LCs were treated with LH and hCG for 12 h and testosterone concentrations in the culture media were then measured. STAC3 deficiency significantly impaired the gonadotropin-induced testosterone synthesis (Fig. 4b). To determine whether transient knockdown of STAC3 also affected testicular steroidogenesis, we tranfected the primary LCs from WT testes with Ctrl shRNA or Stac3 shRNA (Fig. 4c). Subsequent hormone assays revealed that transient knockdown of STAC3 also attenuated the gonadotropin-induced testosterone synthesis in LCs (Fig. 4d). Thus, endogenous expression of STAC3 is required for steroidogenesis in LCs. To further study the functional details of STAC3, we generated the TM3Stac3−/− cells (Fig. 4e). The resultant TM3Stac3−/− cells were stimulated for 12 h with 100 ng/ml LH. Apparently, STAC3 depletion caused a ~ 46.7% decrease in the testosterone secretion in the LH-stimulated cells, but had no effects on the basal testosterone secretion in vitro (Fig. 4f). To identify at which step the steroidogenic process was altered by STAC3 deficiency, TM3Stac3−/− cells were stimulated with different stimulus including db-cAMP, 22-ROH or pregnenolone. ELISA assays using cell culture supernatants showed that testosterone secretion was significantly reduced in db-cAMP- or 22-ROH-treated TM3Stac3−/− cells, but remained unaffected in pregnenolone-treated TM3Stac3−/− cells (Fig. 4g). STAC3 depletion thus may sabotage steroidogenesis by inducing mitochondrial dysfunction.

Effects of inhibition of STAC3 on basal and LH-stimulated testosterone production in primary LCs and TM3 cells. (a) Primary LCs were isolated and purified from Ctrl sh or Stac3 sh-treated testes at 70 d after microinjection. The inhibition of STAC3 in testis was confirmed using immunoblotting. (b) Primary LCsStac3 sh or Ctrl LCs were treated with 100 ng/ml luteinizing hormone (LH)/human chorionic gonadotropin (hCG) for 12 h, followed by measurement of testosterone concentrations in the culture media using an ELISA method (n = 4, *P < 0.05 and **P < 0.01). (c) Primary LCs isolated from wild-type testis were transfected with Stac3 shRNA or Ctrl shRNA. 48 h later, LCs were treated with 100 ng/ml LH/hCG for 12 h, followed by measurement of testosterone concentrations in the culture media using an ELISA method (n = 4, *P < 0.05 and **P < 0.01). (e) Generation of the TM3Stac3−/− cells was confirmed by immunoblotting. (f) TM3Stac3−/− or control cells were treated with 100 ng/ml luteinizing hormone (LH) for 12 h, followed by measurement of testosterone concentrations in the culture medium using an ELISA method (n = 4, *P < 0.05 and **P < 0.01). (g) Testosterone production stimulated for 12 h by db-cAMP (0.5 mM), 22R-hydroxycholesterol (22-ROH, 5 μM), or pregnenolone (5 μM) in the TM3Stac3−/− or control cells was determined as described. Quantitative values are means ± S.E.M. (n = 4, *P < 0.05 and **P < 0.01)
STAC3 depletion impairs mitochondrial membrane potential and StAR processing in db-cAMP-stimulated LCs
Having establishing the functional link between STAC3 expression and mitochondrial dysfunction, we tried to identify the potential molecular basis underpinning our observations. When we investigated the cellular localization of STAC3 in TM3 cells using double-immunofluorescent cytochemistry, STAC3 was observed to be perfectly overlapped with Mitotracker labeling in the cytoplasm (Fig. 5a, a’a’’ and a’’’). A polarized mitochondrial potential is absolutely necessary for steroidogenic activation in LCs, and the magnitude of ΔΨm can be determined by measuring the uptake of TMRE dye in mitochondria (Allen, et al., 2006). We treated the TM3 cells with 0.5 M of db-cAMP to acutely stimulate steroidogenesis and then subjected these cells to TMRE uptake assay. Treatment with db-cAMP induced a significant increase in the uptake of TMRE fluorescence in naïve or Ctrl shRNA-treated TM3 cells, but failed to stimulate noticeable steroidogenic activity in the TM3Stac3−/− cells (Fig. 5b and c). StAR is a rapidly synthesized transport protein in response to intracellular pulses of cAMP. StAR protein regulates the rate-limiting step in steroidogenesis, i.e. the delivery of cholesterol from the outer (OMM) to the inner (IMM) mitochondrial membrane. (Hiroi, et al., 2004). To ask whether the inhibitory effects of STAC3 deficiency on steroidogenesis occurred at the level of StAR, we measured the changes in StAR mRNA and StAR protein. Treatment with db-cAMP induced the expression of the 3.4-kb precursor form of StAR mRNA as well as the 2.9-kb processed form in naïve or Ctrl shRNA-treated TM3 cells. Consistently, db-cAMP stimulated the expression of the 37-kDa precursor form of StAR protein as well as the 30-kDa processed form in both cells. By contrast, STAC3 depletion significantly decreased the processed forms of StAR mRNA (2.9-kb) and protein (30-kDa) upon db-cAMP challenge (Fig. 5d and e). These data together suggest that endogenous STAC3 in LCs is required for mitochondrial membrane potential and StAR processing in the acute regulation of testosterone biosynthesis.

Effects of inhibition of STAC3 on mitochondrial membrane potential and StAR processing in TM3 cells. (a-a’’’) TM3 cells were labeled with Rhodamine Mitotracker, followed by immunofluorescence staining with an anti-STAC3 antibody. Nuclei were counterstained using DAPI. Localization of STAC3 was determined by confocal microscopy. Yellow color denotes the colocalization of STAC3 and Mitotracker. (b) TM3 cells were cultured to ~ 75% confluency in a 96-well florescence assay plate. After incubation with 0.5 mM db-cAMP for 6 h, cells were incubated in 200 μl of a 50-nM solution of TMRE in serum-free medium for 20 min at 37 °C. Uptake of TMRE was then determined by observing TMRE fluorescence under a fluorescence microscope. (c) Quantification of TMRE fluorescence in Penal (b) using a florescence assay plate reader. Data are represented as mean ± S.E.M. from 5 independent experiments. (d) The TM3 cells with different transfections were treated with 0.5 mM db-cAMP for 6 h, followed by non-radioactive Northern blot analysis to measure the changes in the expression of StAR mRNA. (e) TM3 cells with different transfections were treated with 0.5 mM db-cAMP for 6 h, followed by immunoblotting analysis to assess the changes in the expression of StAR protein. TUBULIN was used as the loading control
Discussion
Testicular steroidogenesis is under a delicate control of a cohort of signals from mitochondria. The changes in the intratesticular environment have been observed to influence the mitochondrial homeostasis and dynamics in testicular steroidogenesis (Bjelic, et al., 2015; Radovic, et al., 2019; Shen, et al., 2016; Strauss, et al., 2009). Nevertheless, the key factors responsible for the modulation of mitochondrial homeostasis in activated LCs remain largely unexplored. In the current study, we tested the hypothesis that STAC3, an adaptor protein that is essential for excitation–contraction coupling and calcium channel function in skeletal muscle (Reinholt, et al., 2013), might be functionally expressed in testis. We have shown that STAC3 was expressed with high selectivity in rodent and human LCs (Fig. 1). In agreement, STAC3 expression was negligible in rat testis after selective withdrawal of LCs by administration of the cytotoxic drug EDS, and reappeared immediately after repopulation of LCs (Fig. 2). Thus, LCs serve as the main source of testicular expression of STAC3.
A previous study using microarray analysis has identified STAC3 as one of the most significantly down-regulated genes in infertile male patients. These findings point to a correlation between deregulated expression of STAC3 and spermatogenic failure (Malcher, et al., 2013). Apparently, we could not rule out the possibility that the pathological changes within seminiferous tubules from infertility patients may be detrimental to STAC3 expression. To address this, we employed an in vivo lentiviral vector–mediated microinjection which has been validated to be successful to achieve ectopic gene transfer in LCs (Kim, et al., 2010; Park, et al., 2013). Our results reveal that inhibition of STAC3 expression significantly impaired male fertility by inducing oligozoospermia and asthenospermia, and this impairment can be ascribed to androgen deficiency (Fig. 3, Table 2). Because the immortalized TM3 cells do not fully recapitulate the behavior of endogenous LCs, we validated the indispensible involvement of STAC3 in testicular steroidogenesis using the in vivo knockdown model with isolated primary LCs as well as in vitro experiments with primary cultured LCs (Fig. 4). It is gradually recognized that motion system and male reproduction are coordinately regulated. For example, osteoblast-derived hormone osteocalcin promotes testosterone production and germ cell survival in a CREB-dependent manner (Oury, et al., 2011). By contrast, body wall muscle-produced SWM-1 negatively regulates sperm activation in C. elegans by targeting TRY-5, a spermiogenesis activator (Chavez, et al., 2018). These studies suggest that both bone and skeleton muscle are endocrine organs, and their remodeling throughout adulthood may be tightly associated with male reproductive processes. Our findings extend these understandings by identifying STAC3 as a novel modulator of testicular steroidogenesis. Of note, the mechanisms whereby STAC3 expression is sabotaged in testis with spermatogenesis arrest remain to be defined, one factor has been so far reported to negatively regulate the expression of STAC3. The cyclic adenosine monophosphate (cAMP) is a well-known second messenger that is important in mediating both basal and hCG-stimulated testosterone release in LCs (Kong, et al., 2017). However, abnormal accumulation of cAMP could significantly inhibit testosterone production (Tsai, et al., 1996). Thus, a proper balance in the production of cAMP is likely involved in normal steroidogenesis. Interestingly, there is increasing evidence that accumulation of cAMP correlates to decreased calcium channel activity during impaired testicular steroidogenesis (Tsai, et al., 1997). In light of the critical role of STAC3 in the regulation of calcium channel function (Campiglio, et al., 2018; Campiglio and Flucher, 2017), it will be of future interest to deconvolute if a cAMP/STAC3/Ca2+ axis is also at play in LCs.
By generating the TM3Stac3−/− cells, we further provided the molecular evidence that STAC3 depletion attenuated mitochondrial membrane potential and StAR processing in the db-cAMP-stimulated LCs (Fig. 5). STAC3 was originally identified as a critical EC coupling regulator that triggers contraction of the sarcomere by converting electrical signals to Ca2+ transients (Nelson, et al., 2013). So it is a logical observation that endogenous STAC3 is indispensible for an intact mitochondrial potential. Moreover, ablation of STAC3 expression decreased the processed form of StAR expression at both mRNA and protein levels (Fig. 5d and e). This observation is intriguing, as compared to the previous findings that that disrupting mitochondria usually leads to posttranscriptional changes in StAR during testicular steroidogenesis (Allen, et al., 2006). It is therefore possible that STAC3 may incorporate a transcriptional network to modulate the dynamics of StAR expression in LCs. In favor of our hypothesis, STAC3 has been shown to inhibit the proliferation of pulmonary artery smooth muscle cells (PASMCs) by promoting the deacetylation of FOXO1 transcription factor or by potentiating the activation of PGC-1α, a master coactivator of mitochondrial biogenesis (Zurlo, et al., 2018). The physiological relevance of the crosstalk between STAC3 and its interacting transcription factors in stimulated LCs is presently under evaluation in our lab.
In conclusion, our data demonstrate that the EC coupling regulator STAC3 is expressed in the mitochondria of testicular LCs. We provide the first evidence for a direct involvement of STAC3 in the control of testosterone production by maintaining mitochondrial membrane potential and StAR processing (Fig. 6). Overall, the available data strongly suggest that testicular STAC3 may function as a novel regulator linking mitochondrial homeostasis and testicular steroidogenesis.

Proposed model of the STAC3 signaling pathway in the regulation of mitochondrial membrane potential, StAR processing and testosterone synthesis in LCs
References
Allen JA, Shankara T, Janus P, Buck S, Diemer T, Hales KH, Hales DB (2006) Energized, polarized, and actively respiring mitochondria are required for acute Leydig cell steroidogenesis. Endocrinology 147:3924–3935
Arakane F, King SR, Du Y, Kallen CB, Walsh LP, Watari H, Stocco DM, Strauss JF 3rd (1997) Phosphorylation of steroidogenic acute regulatory protein (StAR) modulates its steroidogenic activity. J Biol Chem 272:32656–32662
Bjelic MM, Stojkov NJ, Radovic SM, Baburski AZ, Janjic MM, Kostic TS, Andric SA (2015) Prolonged in vivo administration of testosterone-enanthate, the widely used and abused anabolic androgenic steroid, disturbs prolactin and cAMP signaling in Leydig cells of adult rats. J Steroid Biochem Mol Biol 149:58–69
Campiglio M, Coste de Bagneaux P, Ortner NJ, Tuluc P, Van Petegem F, Flucher BE (2018) STAC proteins associate to the IQ domain of CaV1.2 and inhibit calcium-dependent inactivation. Proc Natl Acad Sci U S A 115:1376–1381
Campiglio M, Flucher BE (2017) STAC3 stably interacts through its C1 domain with CaV1.1 in skeletal muscle triads. Sci Rep 7:41003
Chavez DR, Snow AK, Smith JR, Stanfield GM (2018) Soma-germ line interactions and a role for muscle in the regulation of C. elegans sperm motility. Development 145
Dong YS, Hou WG, Li Y, Liu DB, Hao GZ, Zhang HF, Li JC, Zhao J, Zhang S, Liang GB, Li W (2016) Unexpected requirement for a binding partner of the syntaxin family in phagocytosis by murine testicular Sertoli cells. Cell Death Differ 23:787–800
Goldberg RB (1984) Germ cell survival, differentiation, and epididymal transit kinetics in mouse testis subjected to high in vivo levels of testosterone enanthate. Cell Tissue Res 237:337–342
He K, Qu H, Wang H, Zhang S, Qian XH, Li W (2016) Regulated and Functional Expression of the Corepressor MTA3 in Rodent Testis. Endocrinology 157:4400–4410
Hiroi H, Christenson LK, Strauss JF 3rd (2004) Regulation of transcription of the steroidogenic acute regulatory protein (StAR) gene: temporal and spatial changes in transcription factor binding and histone modification. Mol Cell Endocrinol 215:119–126
Holdcraft RW, Braun RE (2004) Androgen receptor function is required in Sertoli cells for the terminal differentiation of haploid spermatids. Development 131:459–467
Horstick EJ, Linsley JW, Dowling JJ, Hauser MA, McDonald KK, Ashley-Koch A, Saint-Amant L, Satish A, Cui WW, Zhou W, Sprague SM, Stamm DS, Powell CM, Speer MC, Franzini-Armstrong C, Hirata H, Kuwada JY (2013) Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nat Commun 4:1952
Kim TS, Choi HS, Ryu BY, Gang GT, Kim SU, Koo DB, Kim JM, Han JH, Park CK, Her S, Lee DS (2010) Real-time in vivo bioluminescence imaging of lentiviral vector-mediated gene transfer in mouse testis. Theriogenology 73:129–138
Kong D, Zhang J, Hou X, Zhang S, Tan J, Chen Y, Yang W, Zeng J, Han Y, Liu X, Xu D, Cai R (2017) Acetamiprid inhibits testosterone synthesis by affecting the mitochondrial function and cytoplasmic adenosine triphosphate production in rat Leydig cells. Biol Reprod 96:254–265
Lee JH, Ahn HJ, Lee SJ, Gye MC, Min CK (2011) Effects of L- and T-type Ca(2)(+) channel blockers on spermatogenesis and steroidogenesis in the prepubertal mouse testis. J Assist Reprod Genet 28:23–30
Li W, Zeng Y, Zhao J, Zhu CJ, Hou WG, Zhang S (2014) Upregulation and nuclear translocation of testicular ghrelin protects differentiating spermatogonia from ionizing radiation injury. Cell Death Dis 5:e1248
Malcher A, Rozwadowska N, Stokowy T, Kolanowski T, Jedrzejczak P, Zietkowiak W, Kurpisz M (2013) Potential biomarkers of nonobstructive azoospermia identified in microarray gene expression analysis. Fertil Steril 100(1686–1694):e1681-1687
Midzak AS, Chen H, Aon MA, Papadopoulos V, Zirkin BR (2011) ATP synthesis, mitochondrial function, and steroid biosynthesis in rodent primary and tumor Leydig cells. Biol Reprod 84:976–985
Nelson BR, Wu F, Liu Y, Anderson DM, McAnally J, Lin W, Cannon SC, Bassel-Duby R, Olson EN (2013) Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+ release and contractility. Proc Natl Acad Sci U S A 110:11881–11886
Oury F, Sumara G, Sumara O, Ferron M, Chang H, Smith CE, Hermo L, Suarez S, Roth BL, Ducy P, Karsenty G (2011) Endocrine regulation of male fertility by the skeleton. Cell 144:796–809
Park SJ, Kim TS, Park CK, Lee SH, Kim JM, Lee KS, Lee IK, Park JW, Lawson MA, Lee DS (2013) hCG-induced endoplasmic reticulum stress triggers apoptosis and reduces steroidogenic enzyme expression through activating transcription factor 6 in Leydig cells of the testis. J Mol Endocrinol 50:151–166
Polster A, Dittmer PJ, Perni S, Bichraoui H, Sather WA, Beam KG (2018) Stac Proteins Suppress Ca(2+)-Dependent Inactivation of Neuronal l-type Ca(2+) Channels. J Neurosci 38:9215–9227
Qian W, Xiaoyi W, Zi Y (2019) Screening and Bioinformatics Analysis of IgA Nephropathy Gene Based on GEO Databases. Biomed Res Int 2019:8794013
Radovic SM, Starovlah IM, Capo I, Miljkovic D, Nef S, Kostic TS, Andric SA (2019) Insulin/IGF1 signaling regulates the mitochondrial biogenesis markers in steroidogenic cells of prepubertal testis, but not ovary. Biol Reprod 100:253–267
Reinholt BM, Ge X, Cong X, Gerrard DE, Jiang H (2013) Stac3 is a novel regulator of skeletal muscle development in mice. PLoS ONE 8:e62760
Riccetti L, De Pascali F, Gilioli L, Poti F, Giva LB, Marino M, Tagliavini S, Trenti T, Fanelli F, Mezzullo M, Pagotto U, Simoni M, Casarini L (2017) Human LH and hCG stimulate differently the early signalling pathways but result in equal testosterone synthesis in mouse Leydig cells in vitro. Reprod Biol Endocrinol 15:2
Schoonen M, Smuts I, Louw R, Elson JL, van Dyk E, Jonck LM, Rodenburg RJT, van der Westhuizen FH (2019) Panel-Based Nuclear and Mitochondrial Next-Generation Sequencing Outcomes of an Ethnically Diverse Pediatric Patient Cohort with Mitochondrial Disease. J Mol Diagn 21:503–513
Shen WJ, Azhar S, Kraemer FB (2016) Lipid droplets and steroidogenic cells. Exp Cell Res 340:209–214
Stocco DM, Clark BJ (1996) Role of the steroidogenic acute regulatory protein (StAR) in steroidogenesis. Biochem Pharmacol 51:197–205
Strauss L, Kallio J, Desai N, Pakarinen P, Miettinen T, Gylling H, Albrecht M, Makela S, Mayerhofer A, Poutanen M (2009) Increased exposure to estrogens disturbs maturation, steroidogenesis, and cholesterol homeostasis via estrogen receptor alpha in adult mouse Leydig cells. Endocrinology 150:2865–2872
Sun H, Yang B, Zhu C, Liu R, Wang H, Li W (2013) Presence of metastasis-associated protein 1 in Sertoli cells is required for proper contact between Sertoli cells and adjacent germ cells. Urology 81:66–73
Tena-Sempere M, Barreiro ML, Gonzalez LC, Gaytan F, Zhang FP, Caminos JE, Pinilla L, Casanueva FF, Dieguez C, Aguilar E (2002) Novel expression and functional role of ghrelin in rat testis. Endocrinology 143:717–725
Tian F, Wu YS, Zhao J, Li W (2014) AR3 messenger ribonucleic acid expression and its functional implication in human primary testicular failure. Andrologia 46:859–866
Tsai SC, Chen JJ, Chiao YC, Lu CC, Lin H, Yeh JY, Lo MJ, Kau MM, Wang SW, Wang PS (1997) The role of cyclic AMP production, calcium channel activation and enzyme activities in the inhibition of testosterone secretion by amphetamine. Br J Pharmacol 122:949–955
Tsai SC, Chiao YC, Lu CC, Doong ML, Chen YH, Shih HC, Liaw C, Wang SW, Wang PS (1996) Inhibition by amphetamine of testosterone secretion through a mechanism involving an increase of cyclic AMP production in rat testes. Br J Pharmacol 118:984–988
Wang RS, Yeh S, Chen LM, Lin HY, Zhang C, Ni J, Wu CC, di Sant’Agnese PA, deMesy-Bentley KL, Tzeng CR, Chang C (2006) Androgen receptor in sertoli cell is essential for germ cell nursery and junctional complex formation in mouse testes. Endocrinology 147:5624–5633
Wang X, Zou Z, Yang Z, Jiang S, Lu Y, Wang D, Dong Z, Xu S, Zhu L (2018) HIF 1 inhibits StAR transcription and testosterone synthesis in murine Leydig cells. J Mol Endocrinol 62:1–13
Wu F, Quinonez M, DiFranco M, Cannon SC (2018) Stac3 enhances expression of human CaV1.1 in Xenopus oocytes and reveals gating pore currents in HypoPP mutant channels. J Gen Physiol 150:475–489
Zirkin BR, Papadopoulos V (2018) Leydig cells: formation, function, and regulation. Biol Reprod 99:101–111
Zurlo G, Piquereau J, Moulin M, Pires Da Silva J, Gressette M, Ranchoux B, Garnier A, Ventura-Clapier R, Fadel E, Humbert M, Lemaire C, Perros F, Veksler V (2018) Sirtuin 1 regulates pulmonary artery smooth muscle cell proliferation: role in pulmonary arterial hypertension. J Hypertens 36:1164–1177
Acknowledgments
We wish to express our sincere appreciation to the editor and the anonymous reviewers for their in-depth comments, suggestions, and corrections along the whole editorial process.
Funding
This study was supported by National Key Research and Development Program (Grant No. 2018YFC1002103).
Author information
Authors and Affiliations
Contributions
WX designed and supervised the experiments, contributed reagents, wrote and revised paper and completed the submission. BX completed the majority of experiments, analyzed the data and helped to write the manuscript. LJ and XS performed the experiments and helped to analyze the data. WY contributed partial reagents and helped to analyzed the data.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical approval
The procedures involved in human study, strictly conformed to the 1964 Helsinki Declaration, were approved by the Human Research Committee of Children’s Hospital of Shanxi and Women Health Center of Shanxi (Approval #: CHSX-2014-00164b). All procedures involved in animal work, in compliance with the Guidelines for the Care and Use of Experimental Animals from NIH, were approved by the Institutional Animal Care and Use Committee of Children’s Hospital of Shanxi and Women Health Center of Shanxi (Approval #: CHSX-2014-00164a).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bi, X., Liu, J., Xu, S. et al. Testicular STAC3 regulates Leydig cell steroidogenesis through potentiating mitochondrial membrane potential and StAR processing. Cell Tissue Res 384, 195–209 (2021). https://doi.org/10.1007/s00441-020-03312-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-020-03312-8