
Abstract
CLCN4-related disorder is a rare X-linked neurodevelopmental condition with a pathogenic mechanism yet to be elucidated. CLCN4 encodes the vesicular 2Cl−/H+ exchanger ClC-4, and CLCN4 pathogenic variants frequently result in altered ClC-4 transport activity. The precise cellular and molecular function of ClC-4 remains unknown; however, together with ClC-3, ClC-4 is thought to have a role in the ion homeostasis of endosomes and intracellular trafficking. We reviewed our research database for patients with CLCN4 variants and epilepsy, and performed thorough phenotyping. We examined the functional properties of the variants in mammalian cells using patch-clamp electrophysiology, protein biochemistry, and confocal fluorescence microscopy. Three male patients with developmental and epileptic encephalopathy were identified, with differing phenotypes. Patients #1 and #2 had normal growth parameters and normal-appearing brains on MRI, while patient #3 had microcephaly, microsomia, complete agenesis of the corpus callosum and cerebellar and brainstem hypoplasia. The p.(Gly342Arg) variant of patient #1 significantly impaired ClC-4’s heterodimerization capability with ClC-3 and suppressed anion currents. The p.(Ile549Leu) variant of patient #2 and p.(Asp89Asn) variant of patient #3 both shift the voltage dependency of transport activation by 20 mV to more hyperpolarizing potentials, relative to the wild-type, with p.(Asp89Asn) favouring higher transport activity. We concluded that p.(Gly342Arg) carried by patient #1 and the p.(Ile549Leu) expressed by patient #2 impair ClC-4 transport function, while the p.(Asp89Asn) variant results in a gain-of-transport function; all three variants result in epilepsy and global developmental impairment, but with differences in epilepsy presentation, growth parameters, and presence or absence of brain malformations.





Similar content being viewed by others
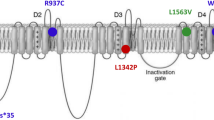
Data availability
No datasets were generated or analysed during the current study.
References
Alekov AK, Fahlke C (2009) Channel-like slippage modes in the human anion/proton exchanger ClC-4. J Gen Physiol 133(5):485–496. https://doi.org/10.1085/jgp.200810155
Andreasen M, Nedergaard S (1996) Dendritic electrogenesis in rat hippocampal CA1 pyramidal neurons: functional aspects of na + and Ca2 + currents in apical dendrites. Hippocampus 6(1):79–95. https://doi.org/10.1002/(SICI)1098-1063(1996)6:1%3C79::AID-HIPO13%3E3.0.CO;2-H
Armstrong CM, Bezanilla F (1974) Charge movement associated with the opening and closing of the activation gates of the na channels. J Gen Physiol 63(5):533–552. https://doi.org/10.1085/jgp.63.5.533
Bolte S, Cordelieres FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224(Pt 3):213–232. https://doi.org/10.1111/j.1365-2818.2006.01706.x
Evans R, O’Neill M, Pritzel A, Antropova N, Senior A, Green T, Žídek A, Bates R, Blackwell S, Yim J, Ronneberger O, Bodenstein S, Zielinski M, Bridgland A, Potapenko A, Cowie A, Tunyasuvunakool K, Jain R, Clancy E, Kohli P, Jumper J, Hassabis D (2022) Protein complex prediction with AlphaFold-Multimer. bioRxiv 202120102004463034. https://doi.org/10.1101/2021.10.04.463034
Friedrich T, Breiderhoff T, Jentsch TJ (1999) Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J Biol Chem 274(2):896–902. https://doi.org/10.1074/jbc.274.2.896
Gentzsch M, Cui L, Mengos A, Chang XB, Chen JH, Riordan JR (2003) The PDZ-binding chloride channel ClC-3B localizes to the Golgi and associates with cystic fibrosis transmembrane conductance regulator-interacting PDZ proteins. J Biol Chem 278(8):6440–6449. https://doi.org/10.1074/jbc.M211050200
Grieschat M, Alekov AK (2012) Glutamate 268 regulates transport probability of the anion/proton exchanger ClC-5. J Biol Chem 287(11):8101–8109. https://doi.org/10.1074/jbc.M111.298265
Guzman RE, Grieschat M, Fahlke C, Alekov AK (2013) ClC-3 is an intracellular chloride/proton exchanger with large voltage-dependent nonlinear capacitance. ACS Chem Neurosci 4(6):994–1003. https://doi.org/10.1021/cn400032z
Guzman RE, Miranda-Laferte E, Franzen A, Fahlke C (2015) Neuronal ClC-3 splice variants differ in subcellular localizations, but mediate identical transport functions. J Biol Chem 290(43):25851–25862
Guzman RE, Bungert-Plumke S, Franzen A, Fahlke C (2017) Preferential association with ClC-3 permits sorting of ClC-4 into endosomal compartments. J Biol Chem 292(46):19055–19065. https://doi.org/10.1074/jbc.M117.801951
Guzman RE, Sierra-Marquez J, Bungert-Plumke S, Franzen A, Fahlke C (2022) Functional characterization of CLCN4 variants Associated with X-Linked intellectual disability and Epilepsy. Front Mol Neurosci 15:872407. https://doi.org/10.3389/fnmol.2022.872407
He H, Guzman RE, Cao D, Sierra-Marquez J, Yin F, Fahlke C, Peng J, Stauber T (2021) The molecular and phenotypic spectrum of CLCN4-related epilepsy. Epilepsia 62(6):1401–1415. https://doi.org/10.1111/epi.16906
Hu H, Haas SA, Chelly J, Van Esch H, Raynaud M, de Brouwer AP, Weinert S, Froyen G, Frints SG, Laumonnier F, Zemojtel T, Love MI, Richard H, Emde AK, Bienek M, Jensen C, Hambrock M, Fischer U, Langnick C, Feldkamp M, Wissink-Lindhout W, Lebrun N, Castelnau L, Rucci J, Montjean R, Dorseuil O, Billuart P, Stuhlmann T, Shaw M, Corbett MA, Gardner A, Willis-Owen S, Tan C, Friend KL, Belet S, van Roozendaal KE, Jimenez-Pocquet M, Moizard MP, Ronce N, Sun R, O’Keeffe S, Chenna R, van Bommel A, Goke J, Hackett A, Field M, Christie L, Boyle J, Haan E, Nelson J, Turner G, Baynam G, Gillessen-Kaesbach G, Muller U, Steinberger D, Budny B, Badura-Stronka M, Latos-Bielenska A, Ousager LB, Wieacker P, Rodriguez Criado G, Bondeson ML, Anneren G, Dufke A, Cohen M, Van Maldergem L, Vincent-Delorme C, Echenne B, Simon-Bouy B, Kleefstra T, Willemsen M, Fryns JP, Devriendt K, Ullmann R, Vingron M, Wrogemann K, Wienker TF, Tzschach A, van Bokhoven H, Gecz J, Jentsch TJ, Chen W, Ropers HH, Kalscheuer VM (2016) X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol Psychiatry 21(1):133–148. https://doi.org/10.1038/mp.2014.193
Ishida Y, Nayak S, Mindell JA, Grabe M (2013) A model of lysosomal pH regulation. J Gen Physiol 141(6):705–720. https://doi.org/10.1085/jgp.201210930
Jentsch TJ (2008) CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit Rev Biochem Mol Biol 43(1):3–36. https://doi.org/10.1080/10409230701829110
Komendantov AO, Ascoli GA (2009) Dendritic excitability and neuronal morphology as determinants of synaptic efficacy. J Neurophysiol 101(4):1847–1866. https://doi.org/10.1152/jn.01235.2007
Kulkarni VA, Firestein BL (2012) The dendritic tree and brain disorders. Mol Cell Neurosci 50(1):10–20. https://doi.org/10.1016/j.mcn.2012.03.005
Lam Z, Wall E, Ryan G, Barber R, Kilby MD, Williams DK (2023) Prenatal diagnosis of CLCN4-related neurodevelopmental disorder in fetuses with congenital brain anomalies. Prenat Diagn 43(9):1247–1250. https://doi.org/10.1002/pd.6404
Li S, Zhang W, Liang P, Zhu M, Zheng B, Zhou W, Wang C, Zhao X (2023) Novel variants in the CLCN4 gene associated with syndromic X-linked intellectual disability. Front Neurol 14:1096969. https://doi.org/10.3389/fneur.2023.1096969
Mohammad-Panah R (2003) The chloride channel ClC-4 contributes to endosomal acidification and trafficking. J Biol Chem
Palmer EE, Stuhlmann T, Weinert S, Haan E, Van Esch H, Holvoet M, Boyle J, Leffler M, Raynaud M, Moraine C, van Bokhoven H, Kleefstra T, Kahrizi K, Najmabadi H, Ropers HH, Delgado MR, Sirsi D, Golla S, Sommer A, Pietryga MP, Chung WK, Wynn J, Rohena L, Bernardo E, Hamlin D, Faux BM, Grange DK, Manwaring L, Tolmie J, Joss S, Study DDD, Cobben JM, Duijkers FAM, Goehringer JM, Challman TD, Hennig F, Fischer U, Grimme A, Suckow V, Musante L, Nicholl J, Shaw M, Lodh SP, Niu Z, Rosenfeld JA, Stankiewicz P, Jentsch TJ, Gecz J, Field M, Kalscheuer VM (2018) De novo and inherited mutations in the X-linked gene CLCN4 are associated with syndromic intellectual disability and behavior and seizure disorders in males and females. Mol Psychiatry 23(2):222–230. https://doi.org/10.1038/mp.2016.135
Palmer EE, Pusch M, Picollo A, Forwood C, Nguyen MH, Suckow V, Gibbons J, Hoff A, Sigfrid L, Megarbane A, Nizon M, Cogne B, Beneteau C, Alkuraya FS, Chedrawi A, Hashem MO, Stamberger H, Weckhuysen S, Vanlander A, Ceulemans B, Rajagopalan S, Nunn K, Arpin S, Raynaud M, Motter CS, Ward-Melver C, Janssens K, Meuwissen M, Beysen D, Dikow N, Grimmel M, Haack TB, Clement E, McTague A, Hunt D, Townshend S, Ward M, Richards LJ, Simons C, Costain G, Dupuis L, Mendoza-Londono R, Dudding-Byth T, Boyle J, Saunders C, Fleming E, El Chehadeh S, Spitz MA, Piton A, Gerard B, Abi Warde MT, Rea G, McKenna C, Douzgou S, Banka S, Akman C, Bain JM, Sands TT, Wilson GN, Silvertooth EJ, Miller L, Lederer D, Sachdev R, Macintosh R, Monestier O, Karadurmus D, Collins F, Carter M, Rohena L, Willemsen MH, Ockeloen CW, Pfundt R, Kroft SD, Field M, Laranjeira FER, Fortuna AM, Soares AR, Michaud V, Naudion S, Golla S, Weaver DD, Bird LM, Friedman J, Clowes V, Joss S, Polsler L, Campeau PM, Blazo M, Bijlsma EK, Rosenfeld JA, Beetz C, Powis Z, McWalter K, Brandt T, Torti E, Mathot M, Mohammad SS, Armstrong R, Kalscheuer VM (2023) Functional and clinical studies reveal pathophysiological complexity of CLCN4-related neurodevelopmental condition. Mol Psychiatry 28(2):668–697. https://doi.org/10.1038/s41380-022-01852-9
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7):676–682. https://doi.org/10.1038/nmeth.2019
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9(7):671–675. https://doi.org/10.1038/nmeth.2089
Sierra-Marquez J, Willuweit A, Schoneck M, Bungert-Plumke S, Gehlen J, Balduin C, Muller F, Lampert A, Fahlke C, Guzman RE (2022) ClC-3 regulates the excitability of nociceptive neurons and is involved in inflammatory processes within the spinal sensory pathway. Front Cell Neurosci 16:920075. https://doi.org/10.3389/fncel.2022.920075
Stauber T, Jentsch TJ (2013) Chloride in vesicular trafficking and function. Annu Rev Physiol 75:453–477. https://doi.org/10.1146/annurev-physiol-030212-183702
Suzuki T, Rai T, Hayama A, Sohara E, Suda S, Itoh T, Sasaki S, Uchida S (2006) Intracellular localization of ClC chloride channels and their ability to form hetero-oligomers. J Cell Physiol 206(3):792–798. https://doi.org/10.1002/jcp.20516
Tian W, Peng LX, Zhao MD, Tao L, Zou P, Zhang Y (2022) Dendritic morphology affects the velocity and amplitude of back-propagating action potentials. Neurosci Bull 38(11):1330–1346. https://doi.org/10.1007/s12264-022-00931-9
van der Velden L, van Hooft JA, Chameau P (2012) Altered dendritic complexity affects firing properties of cortical layer 2/3 pyramidal neurons in mice lacking the 5-HT3A receptor. J Neurophysiol 108(5):1521–1528. https://doi.org/10.1152/jn.00829.2011
Veeramah KR, Johnstone L, Karafet TM, Wolf D, Sprissler R, Salogiannis J, Barth-Maron A, Greenberg ME, Stuhlmann T, Weinert S, Jentsch TJ, Pazzi M, Restifo LL, Talwar D, Erickson RP, Hammer MF (2013a) Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia https://doi.org/10.1111/epi.12201
Weinert S, Gimber N, Deuschel D, Stuhlmann T, Puchkov D, Farsi Z, Ludwig CF, Novarino G, Lopez-Cayuqueo KI, Planells-Cases R, Jentsch TJ (2020) Uncoupling endosomal CLC chloride/proton exchange causes severe neurodegeneration. EMBO J 39(9):e103358. https://doi.org/10.15252/embj.2019103358
Wittig I, Karas M, Schagger H (2007) High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol Cell Proteom 6(7):1215–1225. https://doi.org/10.1074/mcp.M700076-MCP200
Xu X, Lu F, Zhang L, Li H, Du S, Tang J (2021) Novel CLCN4 variant associated with syndromic X-linked intellectual disability in a Chinese girl: a case report. BMC Pediatr 21(1):384. https://doi.org/10.1186/s12887-021-02860-4
Zhou P, He N, Zhang JW, Lin ZJ, Wang J, Yan LM, Meng H, Tang B, Li BM, Liu XR, Shi YW, Zhai QX, Yi YH, Liao WP (2018) Novel mutations and phenotypes of epilepsy-associated genes in epileptic encephalopathies. Genes Brain Behav 17(8):e12456. https://doi.org/10.1111/gbb.12456
Acknowledgements
This study was supported by funding from the Fonds de Recherches du Québec – Santé (KAM) and the German Research Foundation (DFG) (GU 2042/2–1) (REG).
Funding
This study was supported by funding from the Fonds de Recherches du Québec – Santé (KAM) and the German Research Foundation (DFG) (GU 2042/2–1) (REG). The authors have no relevant financial or non-financial interests to disclose.
Author information
Authors and Affiliations
Contributions
Both K.A.M and R.E.G. contributed to the study conception and design. All authors were involved in at least one of material preparation, data collection or analysis. The first draft of the manuscript was written by A.N.S. and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors have no competing interests to report.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sahly, A.N., Sierra-Marquez, J., Bungert-Plümke, S. et al. Genotype-phenotype correlation in CLCN4-related developmental and epileptic encephalopathy. Hum. Genet. (2024). https://doi.org/10.1007/s00439-024-02668-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00439-024-02668-z