
Abstract
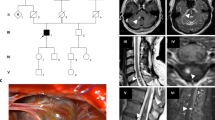
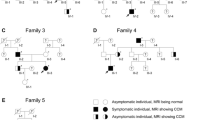
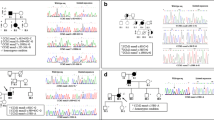
Cerebral cavernous malformations (CCM) are vascular malformations consisting of collections of enlarged capillaries occurring in the brain or spinal cord. These vascular malformations can occur sporadically or susceptibility to develop these can be inherited as an autosomal dominant trait due to mutation in one of three genes. Over a decade ago, we described a 77.6 Kb germline deletion spanning exons 2–10 in the CCM2 gene found in multiple affected individuals from seemingly unrelated families. Segregation analysis using linked, microsatellite markers indicated that this deletion may have arisen at least twice independently. In the ensuing decades, many more CCM patients have been identified with this deletion. In this present study we examined 27 reportedly unrelated affected individuals with this deletion. To investigate the origin of the deletion at base pair level resolution, we sequenced approximately 10 Kb upstream and downstream from the recombination junction on the deleted allele. All patients showed the identical SNP haplotype across this combined 20 Kb interval. In parallel, genealogical records have traced 11 of these individuals to five separate pedigrees dating as far back as the 1600-1700s. These haplotype and genealogical data suggest that these families and the remaining “unrelated” samples converge on a common ancestor due to a founder mutation occurring centuries ago on the North American continent. We also note that another gene, NACAD, is included in this deletion. Although patient self-reporting does not indicate an apparent phenotypic consequence for heterozygous deletion of NACAD, further investigation is warranted for these patients.




Similar content being viewed by others
References
Al-Shahi Salman R, Hall JM, Horne MA et al (2012) Untreated clinical course of cerebral cavernous malformations: a prospective, population-based cohort study. Lancet Neurol 11:217–224. https://doi.org/10.1016/S1474-4422(12)70004-2
Batzer MA, Deininger PL (2002) Alu repeats and human genomic diversity. Nat Rev Genet 3:370–379. https://doi.org/10.1038/nrg798
Bergametti F, Denier C, Labauge P et al (2005) Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet 76:42–51
Booth KT, Azaiez H, Smith RJH (2020) DFNA5 (GSDME) c.991-15_991-13delTTC: founder mutation or mutational hotspot? Int J Mol Sci. https://doi.org/10.3390/ijms21113951
Browning SR, Browning BL (2012) Identity by descent between distant relatives: detection and applications. Annu Rev Genet 46:617–633. https://doi.org/10.1146/annurev-genet-110711-155534
Cau M, Loi M, Melis M, Congiu R, Loi A, Meloni C, Serrenti M, Addis M, Melis MA (2009) C329X in KRIT1 is a founder mutation among CCM patients in Sardinia. Eur J Med Genet 52:344–348. https://doi.org/10.1016/j.ejmg.2009.05.002
Chen MH, Raffield LM, Mousas A et al (2020) Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell 182:1198-213e14. https://doi.org/10.1016/j.cell.2020.06.045
Denier C, Goutagny S, Labauge P et al (2004) Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet 74:326–337
Gallione CJ, Solatycki A, Awad IA, Weber JL, Marchuk DA (2011) A founder mutation in the Ashkenazi Jewish population affecting messenger RNA splicing of the CCM2 gene causes cerebral cavernous malformations. Genet Med 13:662–666. https://doi.org/10.1097/GIM.0b013e318211ff8b
Genealogy Standards (2019) Turner Publishing Company, Nashville
Horne MA, Flemming KD, Su IC et al (2016) Clinical course of untreated cerebral cavernous malformations: a meta-analysis of individual patient data. Lancet Neurol 15:166–173. https://doi.org/10.1016/S1474-4422(15)00303-8
Huyghe JR, Bien SA, Harrison TA et al (2019) Discovery of common and rare genetic risk variants for colorectal cancer. Nat Genet 51:76–87. https://doi.org/10.1038/s41588-018-0286-6
Kichaev G, Bhatia G, Loh PR, Gazal S, Burch K, Freund MK, Schoech A, Pasaniuc B, Price AL (2019) leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet 104:65–75. https://doi.org/10.1016/j.ajhg.2018.11.008
Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF, Tournier-Lasserve E (1999) Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet 23:189–193
Liquori CL, Berg MJ, Siegel AM et al (2003) Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet 73:1459–1464
Liquori CL, Berg MJ, Squitieri F, Leedom TP, Ptacek L, Johnson EW, Marchuk DA (2007) Deletions in CCM2 are a common cause of cerebral cavernous malformations. Am J Hum Genet 80:69–75
Morrison L, and Akers A (1993) Cerebral cavernous malformation, familial. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G , Amemiya G (eds). GeneReviews((R)), Seattle
Reddy R, Efimenko I, Chertman W, Kohn T, Diaz P, Seetharam D, Khodamoradi K, Kresch E, Ramasamy R (2021) Whole exome sequencing identifies a rare mutation in NACAD as a possible cause of COVID orchitis in brothers. Urology. https://doi.org/10.1016/j.urology.2021.09.021
Sahoo T, Johnson EW, Thomas JW et al (1999) Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet 8:2325–2333
Sakaue S, Kanai M, Tanigawa Y et al (2021) A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet 53:1415–1424. https://doi.org/10.1038/s41588-021-00931-x
Song X, Beck CR, Du R et al (2018) Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Res 28:1228–1242. https://doi.org/10.1101/gr.229401.117
Tomsic J, Liyanarachchi S, Hampel H et al (2012) An American founder mutation in MLH1. Int J Cancer 130:2088–2095. https://doi.org/10.1002/ijc.26233
von Salome J, Boonstra PS, Karimi M, Silander G, Stenmark-Askmalm M, Gebre-Medhin S, Aravidis C, Nilbert M, Lindblom A, Lagerstedt-Robinson K (2017) Genetic anticipation in Swedish Lynch syndrome families. PLoS Genet 13:e1007012. https://doi.org/10.1371/journal.pgen.1007012
Vuckovic D, Bao EL, Akbari P et al (2020) The polygenic and monogenic basis of blood traits and diseases. Cell 182:1214-31e11. https://doi.org/10.1016/j.cell.2020.08.008
Zabramski JM, Wascher TM, Spetzler RF, Johnson B, Golfinos J, Drayer BP, Brown B, Rigamonti D, Brown G (1994) The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg 80:422–432
Acknowledgements
We thank the patients who participated in both the genealogical and genetic arms of this study, the patient support group, Angioma Alliance, the CCM2 Exon 2-10 Deletion Families Facebook Group, and our funders: NIH P01-NS092521 (DAM). We also thank Dr. Christine Beck (University of Connecticut Health Center and The Jackson Laboratory for Genomic Medicine) for helpful discussions.
Funding
This work was supported by NIH P01-NS092521 (DAM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gallione, C.J., Detter, M.R., Sheline, A. et al. Genetic genealogy uncovers a founder deletion mutation in the cerebral cavernous malformations 2 gene. Hum Genet 141, 1761–1769 (2022). https://doi.org/10.1007/s00439-022-02458-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-022-02458-5