
Abstract
The anterior pituitary gland secretes several endocrine hormones, essential for growth, reproduction and other basic physiological functions. Abnormal development or function of the pituitary gland leads to isolated or combined pituitary hormone deficiency (CPHD). At least 30 genes have been associated with human CPHD, including many transcription factors, such as POU1F1. CPHD occurs spontaneously also in mice and dogs. Two affected breeds have been reported in dogs: German Shepherds with a splice defect in the LHX3 gene and Karelian Bear Dogs (KBD) with an unknown genetic cause. We obtained samples from five KBDs presenting dwarfism and abnormal coats. A combined analysis of genome-wide association and next-generation sequencing mapped the disease to a region in chromosome 31 and identified a homozygous intronic variant in the fourth exon of the POU1F1 gene in the affected dogs. The identified variant, c.605-3C>A, resided in the splice region and was predicted to affect splicing. The variant's screening in three new prospective cases, related breeds, and ~ 8000 dogs from 207 breeds indicated complete segregation in KBDs with a carrier frequency of 8%, and high breed-specificity as carriers were found at a low frequency only in Lapponian Herders, a related breed. Our study establishes a novel canine model for CPHD with a candidate POU1F1 defect.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The anterior pituitary gland is a critical endocrine organ composed of at least five differentiated cell types and responsible for the secretion of thyroid-stimulating hormone (TSH), growth hormone (GH), prolactin (PRL), adrenocorticotropin (ACTH), follicle-stimulating hormone (FSH) and luteinising hormone (LH) (Kerr et al. 2008; de Moraes et al. 2012). Abnormal development or function of the anterior pituitary gland leads to pituitary hormone deficiency either in an isolated or combined form. Combined pituitary hormone deficiency (CPHD) is characterised by a lack of growth hormone (GH) and at least one additional pituitary hormone. In isolated GH deficiency, the patient is deficient only for GH.
The genetics of CPHD involves at least 30 genes required in the anterior pituitary development, differentiation, and maintenance (Fang et al. 2016; Castinetti et al. 2016), including well-established transcription factor genes PROP1 (Wu et al. 1998), POU1F1 (Tatsumi et al. 1992), HESX1 (Dattani et al. 1998), LHX3 (Netchine et al. 2000), and LHX4 (Machinis et al. 2001). CPHD prevalence is 1:8000, and cases can be categorised into syndromic and non-syndromic groups according to the molecular defects (De Rienzo et al. 2015). A recent genotype screening of the known CPHD genes in ~ 1200 patients revealed only 7.3% and 29.5% discovery rates in sporadic and familial cases, respectively (Jullien et al. 2020). Indeed, the molecular aetiology remains unexplained in most patients despite the accelerating speed of gene discoveries facilitated by high throughput sequencing. Further research is warranted to improve early diagnostics and care since hormone deficiency at birth can result in hypoglycemia and death.
CPHD is spontaneously present also in animals, including mice and dogs. The Snell and Jackson dwarf mice have defective Pou1f1 and lack thyrotroph, somatotroph, and lactotroph development (Bodner et al. 1988; Li et al. 1990). The Ames dwarf mouse with a Prop1 mutation manifests a broader phenotype than the Snell and Jackson mice with an additional lack of gonadotropin levels (LH and FSH) (Sornson et al. 1996). Mouse models have played a fundamental role in understanding pituitary development, function, and disease (Castinetti et al. 2016).
In dogs, pituitary dwarfism has been reported in German Shepherds (Voorbij et al. 2014) and Karelian Bear Dogs (KBD) (Andresen and Willeberg 1976). The affected German Shepherds have a proportionally small stature, a puppy-like (lanugo) coat that lacks most primary or guard hairs, symmetrical alopecia, and a combined deficiency of GH, TSH, and PRL (Allan et al. 1978; Kooistra et al. 2000). The condition is caused by an autosomal recessive 7-bp intronic deletion in LHX3, resulting in defective splicing (Voorbij et al. 2011). The same variant is also present in the related Saarloos and Czechoslovakian Wolfdogs breeds (Voorbij et al. 2014).
Similar to the affected German Shepherds, the affected KBDs also present with growth retardation and coat abnormalities. Andresen and Willeberg (1976) described nine dwarf KBDs from six litters that were approximately 15–20 cm smaller than unaffected KBDs. Some of the affected animals retained their puppy coat, whereas some had a regular appearing adult coat before losing most of it at 2–3 years. Two affected dogs had low somatomedin levels. Pedigree analysis suggested an autosomal recessive disease (Andresen and Willeberg 1976). After 45 years since the original description of the KBD phenotype, we have now identified similar cases from Finland and Sweden and uncovered a candidate causative variant in the POU1F1 gene.
Materials and methods
Study cohorts, pedigree and ethics approval
Canine DNA samples were retrieved from the canine DNA bank at the University of Helsinki, including the KBD sample cohort of 642 dogs utilised in variant discovery and validation. In addition, 263 Lapponian Herders and 87 Laika dogs were used in variant validation. Furthermore, POU1F1 genotype data from 7925 dogs submitted for commercial testing was obtained from Genoscoper Laboratories (Wisdom Health, Helsinki, Finland). We received pedigree information for the affected KBDs from the Finnish Kennel Club’s pedigree registry KoiraNet and a pedigree was drawn using the GenoPro genealogy software. All sampled animals were privately owned pet dogs enrolled in the study with the owner’s informed consent. Sample collection was ethically approved by the Animal Ethics Committee of State Provincial Office of Southern Finland (ESAVI/343/04.10.07/2016 and ESAVI/25696/2020).
Association study
To conduct genome-wide association analysis (GWAS), five affected and 139 unaffected KBDs were array genotyped in four batches with the Illumina CanineHD Beadchip containing 172,963 markers. Before analysis, quality control was performed to remove samples with < 93% call rate and markers with < 95% call rate, < 5% minor allele frequency (MAF), or Hardy–Weinberg equilibrium (HWE) p value < 1 × 10–7 in control dogs. Pruning for call rate was performed before merging of batches and for MAF and HWE after merging. After QC, 144 dogs and 96,274 markers remained. GWAS was performed with plink v1.90b6.10 (Purcell et al. 2007) using basic case–control association analysis (--assoc). P values were corrected for inflation with genomic control correction and for multiple testing with the Bonferroni method. Stratification was evaluated from quantile–quantile and multidimensional scaling plots after analysis. Multidimensional scaling was performed with plink using commands –cluster and –mdsplot 2. Results were visualised using python’s matplotlib module (Hunter 2007).
Next generation sequencing and bioinformatics
Whole genome sequencing (WGS) data from three affected KBDs was produced. Using a 350 bp insert size PCR-free DNA library preparation method, two affected dogs were whole-genome sequenced at 26× and 30× coverage on Illumina high-throughput sequencing platform with a read length of 300 bp (paired-end reads, 2 × 150 bp) at the Novogene Bioinformatics Institute (Beijing, China). Another affected dog was whole-genome sequenced at 14× coverage on HiSeq Illumina sequencing platform with a read length of 252 bp (paired-end reads, 2 × 126 bp) at the University of Bern. The three WGS samples were submitted to the Sequence Read Archive in BAM format with BioProject accession number PRJNA684429 and BioSample accession numbers SAMN17057666, SAMN17057667, and SAMN17057668. The entire next-generation sequencing (NGS) cohort comprised 1038 animals (Online material 1, Suppl Table 1), including WGS data from 804 control animals and whole exome sequence (WES) data from 231 animals. The control cohort was provided by the Dog Biomedical Variant Database Consortium (Jagannathan et al. 2019), and a subset was obtained from the dog and wolf genome SNP database (DoGSD) (Bai et al. 2015).
The reads from the sequenced samples were mapped to the dog reference genome canFam3.1 as described previously (Hytönen et al. 2016). Single nucleotide variant calling was performed as described previously (Hytönen et al. 2016). In addition, mobile-element insertions were detected by MELT (Gardner et al. 2017), and structural variants including deletions, duplications, inversions and insertions by DELLY2 (Rausch et al. 2012). Functional effects of the variants were predicted using ANNOVAR with Ensembl release-100 and NCBI Canis lupus familiaris Annotation Release 105 annotations. The filtering of the single nucleotide variants and small insertions and deletions was performed with the command line version of GQT (Layer et al. 2016), assuming autosomal recessive inheritance. Thus, the affected dogs (case 1 and 4) were required to share the variants in a homozygous state, and controls were allowed to be heterozygous, wild-type, or uncalled. The filtering of the mobile element insertions (MEIs) and structural variants (SVs) was performed using the webGQT variant analysis interface (Arumilli et al. 2020). The analysis was performed with a subset of the whole genome samples under the assumption of autosomal recessive inheritance and with 10 heterozygous animals allowed for the alternative alleles in the control set. All genetic analyses were performed using the dog genome build CanFam 3.1.
Bioinformatic analyses were performed on the prioritised sequence variants obtained from NGS data analysis. Pathogenicity of the missense variants was estimated using the PredictSNP (https://loschmidt.chemi.muni.cz/predictsnp1/) tool, which provides a consensus in silico prediction from several different prediction algorithms (Bendl et al. 2014). The splice region's possible splice effect and synonymous variants were predicted with the NNSPLICE 0.9 tool (Reese et al. 1997). Conservation of the indel and missense variants' positions was assessed using the Multiz 100 vertebrate alignment available in the UCSC Genome Browser. The evolutionary conservation of the POU1F1 splice region variant position was determined by constructing multiple sequence alignment with the Clustal Omega algorithm (https://www.ebi.ac.uk/Tools/msa/clustalo/). The aligned sequences were retrieved from the Entrez protein database (https://www.ncbi.nlm.nih.gov/protein/).
Variant validation
The identified POU1F1 variant was confirmed and validated with Sanger sequencing and Taqman genotyping. Primer3 (http://bioinfo.ut.ee/primer3/) (Rozen and Skaletsky. 2000) was used to design the following Sanger sequencing primers; 5′-CCAGGAAAAGTGTGATCGGG-3′ (forward) and 5′-TCCATCTCCTCTGTACGTTTTG-3′ (reverse). Biotools DNA Polymerase (Biotools B&M Labs, S.A.) was used to perform PCR amplification, Sanger sequencing reactions were performed at the Institute for Molecular Medicine Finland (FIMM), and the obtained Sanger sequence data were analysed with the Sequencher program (Gene Codes Corporation). After initial Sanger confirmation, a custom Taqman SNP genotyping assay (ThermoFisher Scientific) was ordered to genotype the POU1F1 variant in larger sample cohorts. The primer and probe sequences for the assay were the following: 5′-TTTGCATTGTTTTAGAAAGAAAATTTGAAACTCAAA-3′ and 5′-CCTTTTTCTTTCATTTGCTCCCACTT-3′ (forward and reverse, respectively), 5′-VIC-ATTCCCCATTACAGCTTT-3′ and 5′-FAM-CTCACCGTAGTCCCCCAT-3′ (reference and variant allele, respectively). The Taqman genotyping reactions were carried out using Biorad’s CFX96 Touch Real-Time PCR Detection System. The utilised canine POU1F1 reference sequences were NM_001006949.1 and NP_001006950.1 for mRNA and protein, respectively.
Results
Suspected pituitary dwarfism in Karelian Bear Dogs
KBD breeders with possible cases of pituitary dwarfism contacted us to initiate genetic studies. We collected clinical descriptions and blood samples from five dogs, including two Finnish and three Swedish cases. One of the Finnish dogs was a female KBD (case 1) described resembling a 6–7-week-old puppy at 13 weeks of age with a weight of 3.3 kg. At 9.5 months, the dog still weighed only 4 kg, had a “puppy-like” coat, and a height at withers of 34 cm (Fig. 1a, b). As a comparison, adult KBD females and males are typically 49–55 cm high and weigh 17–20 kg, and 54–60 cm tall and weigh 25–28 kg, respectively. A renal ultrasound had been performed for case 1 at the age of 8 weeks due to an increased blood urea nitrogen without abnormalities. Thyroid hormone levels were also within the normal limits at the age of 13 weeks. The two other suspected Finnish KBD cases, an adult female (case 2) (Fig. 1c) and male, were cousins to the mother of case 1 (Fig. 2). Owners reported them to be small with a bad puppy-like coat and hair loss. The female dog (case 2) was only 38 cm high and had substantial bilateral hair-loss affecting the trunk, neck, and proximal limbs (Fig. 1c). The skin of the dog was thin and hyperpigmented in the clinical examination with signs of inflammation. The dog had been treated for hypothyroidism. In Sweden, four suspected pituitary dwarfism cases, two females and two males were reported in a litter of eight puppies. Both of the affected males and one affected female were sampled for this study (cases 3–5) (Fig. 2). The breeder reported small size and abnormal soft coat. The affected puppies weighed 7–10 kg and were 37–40 cm high at the age of 6 months (Fig. 1d). In contrast, the unaffected littermates weighed around 20 kg and were 53–55 cm high. Serum insulin-like growth factor (S-IGF-1) level was measured to be below reference range (83 µg/l, reference 130–180 µg/l) in one affected 6-month-old Swedish dog (case 4), which supported the suspected pituitary dwarfism.

Affected KBDs. a An affected female KBD (case 1) at the age of 9 months. b An affected female dog (case 1) and an unaffected male littermate at 10 months. c An adult affected female KBD (case 2) showing severe hair loss. d An affected KBD puppy from the Swedish litter at the age of 6 months was pictured with a dog from another breed. Photos published with permission from the dog owners under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)

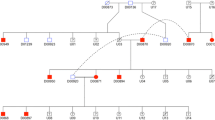
A pedigree drawn around the affected dogs suggests an autosomal recessive condition. The case numbers are denoted as #1, #2 etc. (case 1, case 2). Genotypes for the POU1F1 c.605-3C>A are marked in the pedigree. Cases 1–5 were used in the genome-wide analysis, whereas cases 6–8 were recognised later in the variant validation phase of the study
All the affected dogs were related, both sexes were affected, and parents were unaffected, suggesting an autosomal recessive mode of inheritance (Fig. 2). In addition to the suspected pituitary dwarfism cases, we collected samples from five unaffected full-siblings, four parents, three grandparents, and one great grandparent (Fig. 2).
Disease maps to a locus in chromosome 31
We analysed SNP array data by performing association analysis and homozygosity mapping. Case–control association analysis was carried out with the five affected KBD cases (cases 1–5, Fig. 2) and 139 KBD controls. The analysis revealed two loci that reached a genome-wide significance threshold of 5.19 × 10–7 (Fig. 3). First, fifteen markers were significant in chromosome 31 in a 6.0 Mb region at chr31:2799588–8844745 with the top SNP BICF2S23443199 at 31:4769865 (PGC = 6.83 × 10–15). Second, eleven markers in chromosome 9 were significant in a 3.5 Mb region at chr9:32542006–36040587 with three top SNPs BICF2G630836109, BICF2G630836114 and BICF2G630836121 at 9:34197651 9:34236659 and 9:34275570, respectively (PGC = 1.59 × 10–10). Assessment of genotypes on these two loci revealed all five cases to show extended allelic homozygosity on chromosome 31 (Fig. 3g). In contrast, only four of the five cases showed homozygosity on chromosome 9 (Fig. 3b). Case 2 did not share the same homozygous block. The GWAS results were supported by homozygosity mapping, which revealed regions of homozygous allele sharing in the five affected dogs only on chromosome 31, spanning from 91,801 to 1,792,271 bp and from 4,210,204 to 8,256,918 bp.

Genome-wide association study in five cases and 139 controls. SNP correlation structures (r2) in images b and d were calculated with plink. In images e and g, only ten randomly selected controls are shown for better visualisation. a Manhattan plot. Two genome-wide significant loci were revealed on chromosomes 9 and 31. b A locus plot of chr9:30–38 Mb. c A quantile–quantile plot of raw (UNADJ) and genomic control corrected (GC) p values. Lambda was 1.88 before genomic control correction. d A locus plot of chr31:0–10 Mb. e A genotype plot of chr9:30–38 Mb. One of the top SNPs at chr9:34197651 is indicated with a black triangle. All five cases are above the horizontal line, and ten randomly selected controls below the line. f An MDS plot with two dimensions. Cases are denoted in red and controls in black. g Genotype plot of chr31:0–10 Mb. The candidate variant in POU1F1 is indicated with a red triangle and the top SNP at chr31:4769865 with a black triangle. All five cases are above the horizontal line, and ten randomly selected controls below the line
Whole genome sequencing uncovers a candidate splicing variant in the POU1F1 gene
Whole genome sequencing was performed on three affected KBDs, including two Finnish (cases 1 and 2) and one Swedish dog (case 4) (Fig. 1). Single nucleotide variants and indels obtained from the affected dogs were filtered against 804 control genomes and 231 control exomes (Online material 1, Suppl Table 1) according to an autosomal recessive mode of inheritance. Since case 2 did not share the homozygous block on chromosome 9, it was not regarded as an obligate homozygote in the analysis, in case potential disease-causing variants would be found in chromosome 9. The filtering revealed altogether 321 variants, of which eighteen were not homozygous in case 2 (Online material 2, Suppl Table2). The variants were prioritised according to their position in known protein-coding genes and subsequent potential protein level impact. This approach resulted in the discovery of five exonic variants and one splicing variant (Table 1). All five exonic variants were found on chromosome 9 and comprised two synonymous changes, two missense changes, and one indel called as four separate variants by the analysis pipeline (Table 1). In silico analyses did not predict any splice effect for the synonymous changes, the missense alterations were also predicted be tolerated, and the indel variant and the missense variants to affected non-conserved amino acid positions. Furthermore, these five variants were not present in case 2 and resided in genes with no known direct association to pituitary function or disease. Therefore, they were disregarded as likely disease-causing variants. Instead, all three cases were homozygous for a possible splice variant, c.605-3C>A, located on the fourth intron of the pituitary specific transcription factor gene POU class 1 homeobox 1 (POU1F1), which is composed of altogether six exons. The POU1F1 variant was heterozygous in four control samples (three KBDs and one Lapponian Herder). Variants in the POU1F1 gene cause human CPHD (Tatsumi et al. 1992) and appeared the only suitable causative candidate variant in the affected KBDs. Alignment of the POU1F1 variant position in multiple mammalian species revealed either cytosine or thymine nucleotide in the examined species, suggesting that adenine and guanine might not be tolerated (Online material 3, Suppl Fig. 1). Furthermore, the NNSPLICE 0.9 splice prediction tool (Reese et al. 1997) predicted this variant to weaken the splice acceptor of POU1F1 intron 4 from a score of 0.97 to 0.67.
We also screened a subset of the whole genome samples to identify potential disease-causing larger structural variants (SVs) and mobile element insertions (MEIs). Filtering the three KBD cases against 256 control genomes (Online material 4, Suppl Table 3) uncovered two SVs and three MEIs that were homozygous only in the affected dogs; however, all were either intronic or intergenic changes (Online material 5, Suppl Table 4). One of the three MEIs was located on chromosome 15. One was found outside of the associated region on chromosome 31. The remaining MEI and two SVs were within the associated region on chromosome 31.
Segregation, breed specificity and the carrier frequency of the POUF1F1 variant
We next wanted to evaluate the segregation, breed-specificity, and frequency of the POU1F1 c.605-3C > A variant in dogs. We genotyped the variant in 642 KBDs, including the five affected dogs used in the NGS and SNP chip analyses and all of their available close relatives (Fig. 1). Within the entire KBD sample cohort, 572 dogs were homozygous for the reference allele, 62 were heterozygous, and eight were homozygous for the variant allele (Table 2). The eight KBDs homozygous for the POU1F1 variant comprised the five initially recognised cases from Finland and Sweden and three additional Finnish KBDs. These results indicated a 4% variant allele frequency and an 8% carrier frequency in the Finnish KBD population when the affected dogs and their close relatives (n = 23, Fig. 2) were removed from the calculation.
Two of the new homozygous KBDs (case 6 and 7, Fig. 2) were male puppies from a litter of four males. One puppy from the litter had died during the first day of the life, presumably having been crushed by the dam. The second puppy had stopped eating at the age of 4 days and did not gain weight even with supplementary feeding. The puppy started to have seizures and died at the age of 11 days. The third puppy appeared to develop naturally at first but was found to be blind by a veterinarian, started having movement difficulties, stopped eating and had a seizure-like occurrence. The puppy was euthanised at five weeks of age. The fourth puppy also had problems with sight and was euthanised at the age of 6 weeks. This puppy was sent for autopsy, revealing marked internal hydrocephalus for which no external cause, such as a tumour or infection, could be identified. We obtained samples from the 3rd and 4th puppies euthanised at the ages of 5 and 6 weeks and found them homozygous for the POU1F1 c.605-3C>A variant. The sample for the dam of the litter was not available, but the sire was tested as a carrier. Of note, as the clinical presentation of these two puppies resembled a previously recognized genetic condition occurring in the breed, hypophosphatasia (Kyöstila et al. 2019), the puppies were also tested for the hypophosphatasia-associated ALPL gene variant. Both puppies were homozygous wildtype for the ALPL variant, ruling out this condition. Furthermore, in addition to these two puppies, we obtained a sample from another suspected pituitary dwarfism case (case 8, Fig. 2) from Finland, closely related to the Swedish litter. This female puppy was from a litter of five and was reported to have been of similar size to its littermates when born, but at the age of five weeks was 0.5 kg lighter than other female puppies from the litter, and also had a different coat texture (thinner and longer). This puppy was also genotyped homozygous for the POU1F1 variant.
Since we identified one heterozygous Lapponian Herder (LH) in the NGS analysis, we decided to screen more LHs in our biobank. We genotyped 262 LHs and found seven heterozygotes for the POU1F1 variant, but no homozygotes were identified (Table 2). The cohort of 262 LHs included 29 dogs that were specifically selected from the same breeding lines as the known heterozygous dog, whereas the rest were randomly selected from our biobank. Among the seven new heterozygous dogs, five were from the same breeding line, and two were randomly selected LHs. We also genotyped 87 Laikas from three different varieties (Table 2) to see if the POU1F1 variant is present in a related breed. All Laikas were homozygous wildtype for the POU1F1 variant.
Finally, we reviewed the POU1F1 c.605-3C>A variant genotypes of 7925 dogs from 206 different breeds submitted for genetic testing at Genoscoper Laboratories but found only one heterozygous KBD in this cohort (Online material 6, Suppl table 5).
Discussion
We discovered a homozygous POU1F1 variant in dogs with a clinical phenotype compatible with pituitary dwarfism; a condition reported already 45 years ago in the KBD breed. The association analysis with only five closely related affected KBDs revealed two loci, on chromosome 31 and 9. The latter association did not segregate with the disease in all affected dogs and was likely due to the close relatedness of the cases. However, the locus in chromosome 31 showed complete segregation and included a known candidate gene for pituitary dwarfism, POU1F1, with a splice region variant likely affecting the splice acceptor in exon 4. Our results suggest that similar to humans and mice, POU1F1 underlies pituitary dwarfism also in dogs.
POU1F1, also known as PIT-1, is a late-acting pituitary-specific transcription factor with three functional motifs: an amino-terminal transactivation domain (TAD) and two site-specific DNA binding structures POU-S and POU-H (Andersen and Rosenfeld. 1994). POU1F1 is involved in the differentiation of the somatotroph, lactotroph, and thyrotroph cell lineages and subsequent expression of the GH-1, prolactin and TSH-β genes (Fang et al. 2016). Therefore, POU1F1 gene defects result in GH, PRL, and TSH deficiencies (Tatsumi et al. 1992; Radovick et al. 1992; Aarskog et al. 1997). In addition to CPHD, POU1F1 has been implicated in autosomal dominant isolated GH deficiency (Sobrier et al. 2016). Clinical signs in human POU1F1-related CPHD include severe growth retardation in infancy, proportionate short stature, and distinctive facial features characterised by a prominent forehead, midface hypoplasia, deep-set eyes depressed nasal bridge. Clinical manifestations are present from early life and are associated with a normal or hypoplastic anterior pituitary, while TSH deficiency can be more variable (Radovick et al. 1992; Aarskog et al. 1997; Turton et al. 2005; Fang et al. 2016). The major clinical characteristics in canine pituitary dwarfism are proportionate small stature and an abnormal coat. Low somatomedin values were previously reported in two affected KBDs (Andresen and Willeberg. 1976). In the affected KBDs included in the current study, low serum IGF-1 value had been measured in one dog (case 4), whereas one dog was diagnosed with hypothyroidism (case 2), and one was reported to have normal thyroid hormone values at the age of 13 weeks (case 1).
At least 30 different disease-causing POU1F1 variants have been described to date in humans, of which few are dominant, with the majority showing recessive inheritance (Fang et al. 2016). Our results confirmed autosomal recessive inheritance in the affected KBDs. The POU1F1 splice region variant in affected dogs was predicted to affect splicing. Although functional studies are necessary to confirm the variant's potential splicing effect, it is possible that the variant could result in abnormal splicing, which could, for instance, truncate the C-terminal end of the encoded polypeptide. Unfortunately, we did not have access to the pituitary tissue from affected dogs to test the stability of the mutated transcript and to prove the pathogenicity of the variant. This remains an essential future effort when affected samples become available. Furthermore, the pathogenic effects of the identified variant could be examined through an exon trapping assay. However, there was variation in the clinical presentations of the affected dogs, which could be due to some leakage of the wild-type allele, typical for splice site variants (Hytönen et al. 2016).
We identified the POU1F1 splice region variant in a homozygous state in altogether eight KBDs. The phenotype of six affected dogs (cases 1–5 and 8) was compatible with the clinical description provided by Andresen and Willeberg (1976). Simultaneously, two homozygous male puppies had been euthanised at 5 and 6 weeks of age due to failure to thrive and blindness, suggesting variation in the clinical phenotype. These two puppies also had littermates that died soon after birth without being screened for the POU1F1 variant, and therefore, it is unsure whether they may have also been homozygotes. In German Shephard CPHD, several affected animals were reported to have been intensively nursed and hand-reared during the first three weeks of life due to failure to thrive, which indicated that canine hypopituitarism might go in some instances undetected because of postnatal failure to thrive (Allan et al. 1978).
Despite limitations to validate some of our genetic findings due to lack of appropriate tissue-specific patient material, our study provides compelling evidence to confirm the suspected recessive pituitary dwarfism in KBD, providing a novel comparative large animal model for human CPHD. Simultaneously, the genetic test devised based on this study will have practical implications for improving veterinary diagnostics and breeding plans for better care and eradicating the condition from the affected breeds.
Availability of data and materials
Whole-genome sequencing data from three dogs have been submitted to the Sequence Read Archive in BAM format with BioProject accession number PRJNA684429 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA684429/) and BioSample accession numbers SAMN17057666, SAMN17057667, and SAMN17057668.
Code availability
Not applicable.
References
Aarskog D, Eiken HG, Bjerknes R, Myking OL (1997) Pituitary dwarfism in the R271W Pit-1 gene mutation. Eur J Pediatr 156:829–834. https://doi.org/10.1007/s004310050722
Allan GS, Huxtable CR, Howlett CR, Baxter RC, Duff B, Farrow BR (1978) Pituitary dwarfism in German Shepherd dogs. J Small Anim Pract 19:711–727. https://doi.org/10.1111/j.1748-5827.1978.tb05563.x
Andersen B, Rosenfeld MG (1994) Pit-1 determines cell types during development of the anterior pituitary gland. A model for transcriptional regulation of cell phenotypes in mammalian organogenesis. J Biol Chem 269:29335–29338
Andresen E, Willeberg P (1976) Pituitary dwarfism in Carelian Bear-Dogs: evidence of simple, autosomal recessive inheritance. Hereditas 84:232–234
Arumilli M, Layer RM, Hytönen MK, Lohi H (2020) webGQT: a shiny server for genotype query tools for model-based variant filtering. Front Genet 11:152. https://doi.org/10.3389/fgene.2020.00152
Bai B, Zhao WM, Tang BX, Wang YQ, Wang L, Zhang Z, Yang HC, Liu YH, Zhu JW, Irwin DM, Wang GD, Zhang YP (2015) DoGSD: the dog and wolf genome SNP database. Nucleic Acids Res 43:D777–D783. https://doi.org/10.1093/nar/gku1174
Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, Brezovsky J, Damborsky J (2014) PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol 10:e1003440. https://doi.org/10.1371/journal.pcbi.1003440
Bodner M, Castrillo JL, Theill LE, Deerinck T, Ellisman M, Karin M (1988) The pituitary-specific transcription factor GHF-1 is a homeobox-containing protein. Cell 55:505–518 (pii:0092-8674(88)90037-2)
Castinetti F, Reynaud R, Saveanu A, Jullien N, Quentien MH, Rochette C, Barlier A, Enjalbert A, Brue T (2016) Mechanisms in endocrinology: an update in the genetic aetiologies of combined pituitary hormone deficiency. Eur J Endocrinol 174:R239–R247. https://doi.org/10.1530/EJE-15-1095
Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Martensson IL, Toresson H, Fox M, Wales JK, Hindmarsh PC, Krauss S, Beddington RS, Robinson IC (1998) Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet 19:125–133. https://doi.org/10.1038/477
de Moraes DC, Vaisman M, Conceicao FL, Ortiga-Carvalho TM (2012) Pituitary development: a complex, temporal regulated process dependent on specific transcriptional factors. J Endocrinol 215:239–245. https://doi.org/10.1530/JOE-12-0229
De Rienzo F, Mellone S, Bellone S, Babu D, Fusco I, Prodam F, Petri A, Muniswamy R, De Luca F, Salerno M, Momigliano-Richardi P, Bona G, Giordano M, Italian Study Group on Genetics of CPHD (2015) Frequency of genetic defects in combined pituitary hormone deficiency: a systematic review and analysis of a multicentre Italian cohort. Clin Endocrinol (Oxf) 83:849–860. https://doi.org/10.1111/cen.12849
Fang Q, George AS, Brinkmeier ML, Mortensen AH, Gergics P, Cheung LY, Daly AZ, Ajmal A, Perez Millan MI, Ozel AB, Kitzman JO, Mills RE, Li JZ, Camper SA (2016) Genetics of combined pituitary hormone deficiency: roadmap into the genome era. Endocr Rev 37:636–675. https://doi.org/10.1210/er.2016-1101
Gardner EJ, Lam VK, Harris DN, Chuang NT, Scott EC, Pittard WS, Mills RE, 1000 Genomes Project Consortium, Devine SE (2017) The Mobile Element Locator Tool (MELT): population-scale mobile element discovery and biology. Genome Res 27:1916–1929. https://doi.org/10.1101/gr.218032.116
Hunter JD (2007) Matplotlib: a 2D graphics environment. Comput Sci Eng 9:90–95
Hytönen MK, Arumilli M, Lappalainen AK, Owczarek-Lipska M, Jagannathan V, Hundi S, Salmela E, Venta P, Sarkiala E, Jokinen T, Gorgas D, Kere J, Nieminen P, Drogemuller C, Lohi H (2016) Molecular characterization of three canine models of human rare bone diseases: caffey, van den Ende-Gupta, and raine syndromes. PLoS Genet 12:e1006037. https://doi.org/10.1371/journal.pgen.1006037
Jagannathan V, Drogemuller C, Leeb T, Dog Biomedical Variant Database Consortium (DBVDC) (2019) A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim Genet 50:695–704. https://doi.org/10.1111/age.12834
Jullien N, Saveanu A, Vergier J, Marquant E, Quentien MH, Castinetti F, Galon-Faure N, Brauner R, Turki MZ, Tauber M, El Kholy M, Linglart A, Rodien P, Fedala NS, Bergada I, Cortet-Rudelli C, Polak M, Nicolino M, Stuckens C, Barlier A, Brue T, Reynaud R, Network G (2020) Clinical lessons learned in constitutional hypopituitarism from two decades of experience in a large international cohort. Clin Endocrinol (Oxf). https://doi.org/10.1111/cen.14355
Kerr J, Wood W, Ridgway EC (2008) Basic science and clinical research advances in the pituitary transcription factors: Pit-1 and Prop-1. Curr Opin Endocrinol Diabetes Obes 15:359–363. https://doi.org/10.1097/MED.0b013e3283060a56
Kooistra HS, Voorhout G, Mol JA, Rijnberk A (2000) Combined pituitary hormone deficiency in German shepherd dogs with dwarfism. Domest Anim Endocrinol 19:177–190 (pii: S0739-7240(00)00074-6)
Kyöstila K, Syrja P, Lappalainen AK, Arumilli M, Hundi S, Karkamo V, Viitmaa R, Hytönen MK, Lohi H (2019) A homozygous missense variant in the alkaline phosphatase gene ALPL is associated with a severe form of canine hypophosphatasia. Sci Rep. https://doi.org/10.1038/s41598-018-37801-2
Layer RM, Kindlon N, Karczewski KJ, Exome Aggregation Consortium, Quinlan AR (2016) Efficient genotype compression and analysis of large genetic-variation data sets. Nat Methods 13:63–65. https://doi.org/10.1038/nmeth.3654
Li S, Crenshaw EB 3rd, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG (1990) Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nature 347:528–533. https://doi.org/10.1038/347528a0
Machinis K, Pantel J, Netchine I, Leger J, Camand OJ, Sobrier ML, Dastot-Le Moal F, Duquesnoy P, Abitbol M, Czernichow P, Amselem S (2001) Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet 69:961–968 (pii: S0002-9297(07)61312-3)
Netchine I, Sobrier ML, Krude H, Schnabel D, Maghnie M, Marcos E, Duriez B, Cacheux V, Moers A, Goossens M, Gruters A, Amselem S (2000) Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet 25:182–186. https://doi.org/10.1038/76041
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575. https://doi.org/10.1086/519795
Radovick S, Nations M, Du Y, Berg LA, Weintraub BD, Wondisford FE (1992) A mutation in the POU-homeodomain of Pit-1 responsible for combined pituitary hormone deficiency. Science 257:1115–1118. https://doi.org/10.1126/science.257.5073.1115
Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO (2012) DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28:i333–i339. https://doi.org/10.1093/bioinformatics/bts378
Reese MG, Eeckman FH, Kulp D, Haussler D (1997) Improved splice site detection in Genie. J Comput Biol 4:311–323. https://doi.org/10.1089/cmb.1997.4.311
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Sobrier ML, Tsai YC, Perez C, Leheup B, Bouceba T, Duquesnoy P, Copin B, Sizova D, Penzo A, Stanger BZ, Cooke NE, Liebhaber SA, Amselem S (2016) Functional characterisation of a human POU1F1 mutation associated with isolated growth hormone deficiency: a novel etiology for IGHD. Hum Mol Genet 25:472–483. https://doi.org/10.1093/hmg/ddv486
Sornson MW, Wu W, Dasen JS, Flynn SE, Norman DJ, O’Connell SM, Gukovsky I, Carriere C, Ryan AK, Miller AP, Zuo L, Gleiberman AS, Andersen B, Beamer WG, Rosenfeld MG (1996) Pituitary lineage determination by the Prophet of Pit-1 homeodomain factor defective in Ames dwarfism. Nature 384:327–333. https://doi.org/10.1038/384327a0
Tatsumi K, Miyai K, Notomi T, Kaibe K, Amino N, Mizuno Y, Kohno H (1992) Cretinism with combined hormone deficiency caused by a mutation in the PIT1 gene. Nat Genet 1:56–58. https://doi.org/10.1038/ng0492-56
Turton JP, Reynaud R, Mehta A, Torpiano J, Saveanu A, Woods KS, Tiulpakov A, Zdravkovic V, Hamilton J, Attard-Montalto S, Parascandalo R, Vella C, Clayton PE, Shalet S, Barton J, Brue T, Dattani MT (2005) Novel mutations within the POU1F1 gene associated with variable combined pituitary hormone deficiency. J Clin Endocrinol Metab 90:4762–4770 (pii: jc.2005-0570)
Voorbij AM, van Steenbeek FG, Vos-Loohuis M, Martens EE, Hanson-Nilsson JM, van Oost BA, Kooistra HS, Leegwater PA (2011) A contracted DNA repeat in LHX3 intron 5 is associated with aberrant splicing and pituitary dwarfism in German shepherd dogs. PLoS ONE 6:e27940. https://doi.org/10.1371/journal.pone.0027940
Voorbij AM, Leegwater PA, Kooistra HS (2014) Pituitary dwarfism in Saarloos and Czechoslovakian wolfdogs is associated with a mutation in LHX3. J Vet Intern Med 28:1770–1774. https://doi.org/10.1111/jvim.12448
Wu W, Cogan JD, Pfaffle RW, Dasen JS, Frisch H, O’Connell SM, Flynn SE, Brown MR, Mullis PE, Parks JS, Phillips JA 3rd, Rosenfeld MG (1998) Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet 18:147–149. https://doi.org/10.1038/ng0298-147
Acknowledgements
We would like to acknowledge the Biomedicum Functional Genomics Unit (FuGU) and the Institute for Molecular Medicine Finland core facility (FIMM) at the University of Helsinki for sequencing services, as well as the IT Center for Science Ltd. (CSC, Finland) for providing high-performance computing infrastructure. Sini Karjalainen and Ileana Quintero are thanked for expert technical help. We thank the Dog Biomedical Variant Database Consortium (DBVDC), Gustavo Aguirre, Catherine André, Danika Bannasch, Doreen Becker, Cord Drögemüller, Kari Ekenstedt, Kiterie Faller, Oliver Forman, Steve Friedenberg, Eva Furrow, Urs Giger, Christophe Hitte, Marjo K Hytönen, Vidhya Jagannathan, Frode Lingaas, Tosso Leeb, Hannes Lohi, Cathryn Mellersh, James R Mickelson, Leonardo Murgiano, Anita Oberbauer, Sheila Schmutz, Jeffrey J Schoenebeck, Kim Summers, Frank van Steenbeek and Claire Wade for sharing the control genome sequence data from dogs and wolves. The authors acknowledge the dog owners for providing DNA samples from their dogs.
Funding
Open Access funding provided by University of Helsinki including Helsinki University Central Hospital. This study was partially supported by the Jane and Aatos Erkko Foundation, the Academy of Finland, the Dog Health Research Fund, the Helsinki Institute of Life Science, and Wisdom Health.
Author information
Authors and Affiliations
Contributions
Conceptualisation: HL and KK; Design: KK, HL; Investigation: KK, JEN, MA, MKH; Formal analysis: KK, MKH, JEN, JD; Resources: HL; Writing–original draft: KK and HL; Writing–editing and review: all; Supervision: HL; Funding acquisition: HL.
Corresponding author
Ethics declarations
Conflict of interest
HL is a paid consultant to Genoscoper Laboratories Ltd, providing genetic tests for dogs. JD is an employee of Genoscoper Laboratories Ltd.
Ethics approval
Sample collection was ethically approved by the Animal Ethics Committee of State Provincial Office of Southern Finland (ESAVI/343/04.10.07/2016 and ESAVI/25696/2020).
Consent to participate
Written consent was given by the dogs’ owners, who donated samples from their dogs.
Consent for publication
The owners gave written consent for the dogs’ photos.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kyöstilä, K., Niskanen, J.E., Arumilli, M. et al. Intronic variant in POU1F1 associated with canine pituitary dwarfism. Hum Genet 140, 1553–1562 (2021). https://doi.org/10.1007/s00439-021-02259-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-021-02259-2