
Abstract
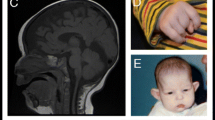
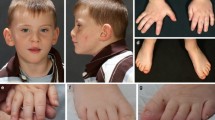
Recently, de novo heterozygous loss-of-function mutations in beta-catenin (CTNNB1) were described for the first time in four individuals with intellectual disability (ID), microcephaly, limited speech and (progressive) spasticity, and functional consequences of CTNNB1 deficiency were characterized in a mouse model. Beta-catenin is a key downstream component of the canonical Wnt signaling pathway. Somatic gain-of-function mutations have already been found in various tumor types, whereas germline loss-of-function mutations in animal models have been shown to influence neuronal development and maturation. We report on 16 additional individuals from 15 families in whom we newly identified de novo loss-of-function CTNNB1 mutations (six nonsense, five frameshift, one missense, two splice mutation, and one whole gene deletion). All patients have ID, motor delay and speech impairment (both mostly severe) and abnormal muscle tone (truncal hypotonia and distal hypertonia/spasticity). The craniofacial phenotype comprised microcephaly (typically −2 to −4 SD) in 12 of 16 and some overlapping facial features in all individuals (broad nasal tip, small alae nasi, long and/or flat philtrum, thin upper lip vermillion). With this detailed phenotypic characterization of 16 additional individuals, we expand and further establish the clinical and mutational spectrum of inactivating CTNNB1 mutations and thereby clinically delineate this new CTNNB1 haploinsufficiency syndrome.


Similar content being viewed by others

References
Campos VE, Du M, Li Y (2004) Increased seizure susceptibility and cortical malformation in β-catenin mutant mice. Biochem Biophys Res Commun 320(2):606–614
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, del Rosario M, Hoischen A, Scheffer H, de Vries BB, Brunner HG, Veltman JA, Vissers LE (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367:1921–1929
Dubruc E, Putoux A, Labalme A, Rougeot C, Sanlaville D, Edery P (2014) A new intellectual disability syndrome caused by CTNNB1 haploinsufficiency. Am J Med Genet A 164:1571–1575
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP (2009) DECIPHER: database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 84:524–533
Flore LA, Milunsky JM (2012) Updates in the genetic evaluation of the child with global developmental delay or intellectual disability. Semin Pediatr Neurol 19:173–180
Kuechler A, Zink AM, Wieland T, Lüdecke H-J, Cremer K, Salviati L, Magini P, Najafi K, Zweier C, Czeschik JC, Aretz S, Endele S, Tamburrino F, Pinato C, Clementi M, Gundlach J, Maylahn C, Mazzanti L, Wohlleber E, Schwarzmayr T, Kariminejad R, Schlessinger A, Wieczorek D, Strom TM, Novarino G, Engels H (2014) Loss-of-function variants of SETD5 cause intellectual disability and the core phenotype of microdeletion 3p25.3 syndrome. Eur J Hum Genet. doi:10.1038/ejhg.2014.165
Lang B, Pu J, Hunter I, Liu M, Martin-Granados C, Reilly TJ, Gao GD, Guan ZL, Li WD, Shi YY, He G, He L, Stefánsson H, St Clair D, Blackwood DH, McCaig CD, Shen S (2014) Recurrent deletions of ULK4 in schizophrenia: a gene crucial for neuritogenesis and neuronal motility. J Cell Sci 127:630–640
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25:1754–1760
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303
O’Roak BJ et al (2012a) Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338(6114):1619–1622
O’Roak BJ et al (2012b) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485(7397):246–250
Rauch A, Wieczorek D, Graf E et al (2012) Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380:1674–1682
Ropers HH (2010) Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet 11:161–187
Tucci V, Kleefstra T, Hardy A, Heise I, Maggi S, Willemsen MH, Hilton H, Esapa C, Simon M, Buenavista MT, McGuffin LJ, Vizor L, Dodero L, Tsaftaris S, Romero R, Nillesen WN, Vissers LE, Kempers MJ, Vulto-van Silfhout AT, Iqbal Z, Orlando M, Maccione A, Lassi G, Farisello P, Contestabile A, Tinarelli F, Nieus T, Raimondi A, Greco B, Cantatore D, Gasparini L, Berdondini L, Bifone A, Gozzi A, Wells S, Nolan PM (2014) Dominant β-catenin mutations cause intellectual disability with recognizable syndromic features. J Clin Invest 124(4):1468–1482. doi:10.1172/JCI70372
Van der Auwera GA, Carneiro M, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella K, Altshuler D, Gabriel S, DePristo M (2013) From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Current Protoc Bioinform 43:11.10.1–11.10.33
Vogel P, Read RW, Hansen GM, Payne BJ, Small D, Sands AT, Zambrowicz BP (2012) Congenital hydrocephalus in genetically engineered mice. Vet Pathol 49:166–181
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164
Xing Y, Takemaru K, Liu J, Berndt JD, Zheng JJ, Moon RT, Xu W (2008) Crystal structure of a full-length beta-catenin. Structure 16:478–487
Yu X, Malenka RC (2003) Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci 6:1169–1177
Acknowledgments
We are grateful to the patients and their families for participating in this study and for giving consent to publish data and photographs. We thank Sabine Kaya and Daniela Falkenstein for excellent technical assistance. This work was supported by grants of The Netherlands Organization for Health Research and Development (ZonMw grant 907-00-365 to TK) and the European Union under the 7th framework program (Gencodys HEALTH-F4-2010-241995 to HvB and TK).
Conflict of interest
The authors declare that they have no competing interests. The views expressed are those of the author and do not reflect the official policy of the Department of the Army, the Department of Defense or the U. S. Government.
Author information
Authors and Affiliations
Corresponding author
Additional information
T. Kleefstra and D. Wieczorek contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
439_2014_1498_MOESM1_ESM.tif
Supplementary figure S1: Cranial MRI scan of patient 8 indicating (A) dysgenesis of the corpus callosum (arrow), hypoplastic brainstem, and (B) enlarged lateral ventricles.(TIFF 520 kb)
439_2014_1498_MOESM2_ESM.pdf
Supplementary Table S1: Detailed clinical data of all patients with CTNNB1 mutations known so far. n.e. not examined, n.r. not recorded, nl normal, PDA persistent ductus arteriosus, SD standard deviations, (l) left, (r) right, (*L published by de Ligt et al, 2012), (*T published by Tucci et al, 2014), (*D published by Dubruc et al, 2014), (** microcephaly of –2.1 SD at age 32 months), (***enlarged lateral ventricles, dysgenesis of the corpus callosum, abnormal gyration of the temporal lobe, absence of the right fornix, hypoplastic brainstem).(PDF 245 kb)
Rights and permissions
About this article

Cite this article
Kuechler, A., Willemsen, M.H., Albrecht, B. et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum Genet 134, 97–109 (2015). https://doi.org/10.1007/s00439-014-1498-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-014-1498-1