
Abstract
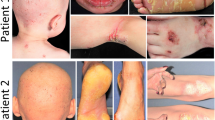
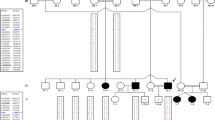
Erythrokeratodermia variabilis 3 (Kamouraska type) or EKV3 is a newly described autosomal recessive disorder observed in patients from the Bas St-Laurent region of Quebec. It has similar skin lesions as observed for EKV, including congenital hyperkeratosis and red patches of variable sizes, shapes, and duration. EKV3 is also characterized by ichthyosis, sensorineural hearing loss, peripheral neuropathy, psychomotor retardation, congenital chronic diarrhea, and an elevation of very long chain fatty acids (VLCFAs). To map the disease locus, we performed candidate gene analysis and a genomewide scan to identify a common homozygous region in affected individuals from three non-consanguineous families. Mutations in connexin 31 (GJB3) and connexin 30.3 (GJB4), implicated in previous reports of EKV, and connexin 26 (GJB2), implicated in palmoplantar keratoderma, were unlikely given the lack of shared homozygous haplotypes in the regions surrounding these genes. The most promising region of common homozygosity observed in a 4,600 single-nucleotide polymorphism genome scan was further characterized by using microsatellites. A 6.8-Mb region on chromosome 7 between D7S2539 and rs727708 was found to be homozygous for the same haplotype in all affected individuals but not in the parents or an unaffected sibling. This region contains connexin 31.3 (GJE1), and although no mutation have been observed in the coding region of this gene, further analyses are required in order to exclude it. Identification of the gene responsible for this disorder will provide insights into the etiology of this multisystemic disorder.


Similar content being viewed by others

References
Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL (2002) Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci 22:6458–6470
Baumgartner MR, Poll-The BT, Verhoeven NM, Jakobs C, Espeel M, Roels F, Rabier D, Levade T, Rolland MO, Martinez M, Wanders RJ, Saudubray JM (1998) Clinical approach to inherited peroxisomal disorders: a series of 27 patients. Ann Neurol 44:720–730
Beare JM, Nevin NC, Froggatt P, Kernohan DC, Allen IV (1972) Atypical erythrokeratoderma with deafness, physical retardation and peripheral neuropathy. Br J Dermatol 87:308–314
Drouin R, Therrien J-P, Angers M, Ouellet S (2001) In vivo DNA analysis. Methods Mol Biol 148:175–219
Engert JC, Vohl MC, Williams SM, Lepage P, Loredo-Osti JC, Faith J, Dore C, Renaud Y, Burtt NP, Villeneuve A, Hirschhorn JN, Altshuler D, Groop LC, Despres JP, Gaudet D, Hudson TJ (2002) 5′ Flanking variants of resistin are associated with obesity. Diabetes 51:1629–1634
Gottfried I, Landau M, Glaser F, Di WL, Ophir J, Mevorah B, Ben-Tal N, Kelsell DP, Avraham KB (2002) A mutation in GJB3 is associated with recessive erythrokeratodermia variabilis (EKV) and leads to defective trafficking of the connexin 31 protein. Hum Mol Genet 11:1311–1316
Itin PH, Moschopulos M, Richard G (2003) Reticular erythrokeratoderma: a new disorder of cornification. Am J Med Genet 120A:237–240
Kruglyak L (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 22:139–144
Leonard AL, Freedberg IM (2003) Palmoplantar keratoderma of Sybert. Dermatol Online J 9:30
Levy J, Chung W, Garzon M, Gallagher MP, Oberfield SE, Lieber E, Anyane-Yeboa K (2003) Congenital myopathy, recurrent secretory diarrhea, bullous eruption of skin, microcephaly, and deafness: a new genetic syndrome? Am J Med Genet 116A:20–25
Lopez-Bigas N, Olive M, Rabionet R, Ben-David O, Martinez-Matos JA, Bravo O, Banchs I, Volpini V, Gasparini P, Avraham KB, Ferrer I, Arbones ML, Estivill X (2001) Connexin 31 (GJB3) is expressed in the peripheral and auditory nerves and causes neuropathy and hearing impairment. Hum Mol Genet 10:947–952
Macari F, Landau M, Cousin P, Mevorah B, Brenner S, Panizzon R, Schorderet DF, Hohl D, Huber M (2000) Mutation in the gene for connexin 30.3 in a family with erythrokeratodermia variabilis. Am J Hum Genet 67:1296–1301
Naidu S, Moser HW (1994) Peroxisomal disorders. Neurol Clin 12:727–739
Oliphant A, Barker DL, Stuelpnagel JR, Chee MS (2002) BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotechniques 32:S56–S61
Plantard L, Huber M, Macari F, Meda P, Hohl D (2003) Molecular interaction of connexin 30.3 and connexin 31 suggests a dominant-negative mechanism associated with erythrokeratodermia variabilis. Hum Mol Genet 12:3287–3294
Rabionet R, Gasparini P, Estivill X (2000) Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Hum Mutat 16:190–202
Richard G, Brown N, Smith LE, Terrinoni A, Melino G, Mackie RM, Bale SJ, Uitto J (2000) The spectrum of mutations in erythrokeratodermias–novel and de novo mutations in GJB3. Hum Genet 106:321–329
Sohl G, Willecke K (2004) Gap junctions and the connexin protein family. Cardiovasc Res 62:228–232
Tremblay S, Khandjian EW (1998) Successful use of long-term frozen lymphocytes for the establishment of lymphoblastoid cell lines. Clin Biochem 31:555–556
Acknowledgements
The authors thank David Rosenblatt for useful advice and are grateful for the technical help given by members of the McGill University and Genome Quebec Innovation Centre, including Corinne Darmond-Zwaig and Carole Doré. The authors are also grateful to Nancy Dallaire, Isabelle Paradis, and Sandra Tremblay for their excellent technical support. This project was supported by the Canadian Genetics and Diseases Network (to T.J.H. and R.D.). R.D. holds the Canada Research Chair in “Genetics, Mutagenesis, and Cancer”. A.M. is supported by a fellowship from the Canadian Institute of Health Research (CIHR).
Author information
Authors and Affiliations
Corresponding author
Additional information
T.G. Saba and A. Montpetit have contributed equally to this work
Rights and permissions
About this article
Cite this article
Saba, T.G., Montpetit, A., Verner, A. et al. An atypical form of erythrokeratodermia variabilis maps to chromosome 7q22. Hum Genet 116, 167–171 (2005). https://doi.org/10.1007/s00439-004-1193-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-004-1193-8