
Abstract
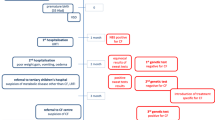
We report on a 6-month-old premature boy from consanguineous parents. He presented with respiratory distress, necrotizing enterocolitis and hyperbilirubinemia shortly after birth. Persisting respiratory symptoms and failure to thrive prompted cystic fibrosis diagnostics, which showed the lack of wild-type signal for the mutation R347P suggesting a homozygous deletion or an alteration different from the known mutation at this position. Sequencing of this region revealed the homozygous substitution 1175 T > A (HGVS: c.1043 T > A) in exon 7 resulting in the homozygous amino acid change M348K. This mutation has never been reported in homozygosity before. Computational analysis tools classified M348K as ‘presumably disease causing.’ In our patient, sweat testing and electrophysiological assessment of CFTR function in native rectal epithelium demonstrated normal Cl− secretion. Conclusion: We assume that the homozygous alteration M348K is a harmless variant rather than a CF-causing mutation.





Similar content being viewed by others


References
Adzhubei L, Schmidt S, Peshkin L, Ramensky V, Gerasimova A, Bork P, Kondrashov A, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249
Audrezet MP, Novelli G, Mercier B, Sangiuolo F, Maceratesi P, Ferec C, Dallapiccola B (1993) Identification of three novel cystic fibrosis mutations in a sample of Italian cystic fibrosis patients. Hum Hered 43:295–300
CF Mutation Database. http://www.genet.sickkids.on.ca
D’Apice MR, Gambardella S, Russo S, Lucidi V, Nardone AM, Pietropolli A, Novelli G (2004) Segregation analysis in cystic fibrosis at-risk family demonstrates that the M348K CFTR mutation is a rare innocuous polymorphism. Prenat Diagn 24:981–983
Deltas CC, Boteva K, Georgiou A, Papageorgiou E, Georgiou C (1996) Description of a symptomless cystic fibrosis L346P/M348K compound heterozygous Cypriot individual. Mol Cell Probes 10:315–318
Ferrer-Costa C, Gelpí JL, Zamakola L, Parraga I, de la Cruz X, Orozco M (2005) PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 21:3176–3178
Hirtz S, Gonska T, Seydewitz HH, Thomas J, Greiner P, Kuehr J, Brandis M, Eichler I, Rocha H, Lopes AI, Barreto C, Ramalho A, Amaral MD, Kunzelmann K, Mall M (2004) CFTR Cl− channel function in native human colon correlates with the genotype and phenotype in cystic fibrosis. Gastroenterology 127:1085–1095
Human Genome Mutation Database. http://www.hgmd.cf.ac.uk
Mall M, Hirtz S, Gonska T, Kunzelmann K (2004) Assessment of CFTR function in rectal biopsies for the diagnosis of cystic fibrosis. J Cyst Fibros 3:165–169
Mall M, Wissner A, Seydewitz H, Hubner MK, Kuelir J, Brandis M, Greger R, Kunzelmann K (2000) Effect of genistein on native epithelial tissue from normal individuals and CF patients and on ion channels expressed in Xenopus oocytes. Br J Pharmacology 130:1884–1892
MutationTaster. http://neurocore.charite.de/MutationTaster/documentation.html
NetGene2. http://www.cbs.dtu.dk/services/NetGene2/
Ng PC, Henikoff S (2003) SIFT: predicting amino acid changes that affect protein function. Nucl Acids Res 31:3812–3814
PolyPhen2. http://genetics.bwh.harvard.edu/pph2/
Pringle I (2002) Studies on the persistence of transgene expression in the murine airways. D.Phil thesis, Oxford
ProteinAtlas. www.proteinatlas.org
Roth EK, Hirtz S, Duerr J, Wenning D, Eichler I, Seydewitz HH, Amaral MD, Mall MA (2011) The K < sup > +</sup > channel opener 1-EBIO potentiates residual function of mutant CFTR in rectal biopsies from cystic fibrosis patients. PLoS One 6:e24445
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat Meth 7:575–576
Sheppard DN, Welsh MJ (1999) Structure and function of the CFTR chloride channel. Physiol Rev 79:S23–S45
SIFT. http://sift.jcvi.org
UCSC. www.genome.ucsc.edu
UniProt. http://www.uniprot.org/uniprot/
World Health Organisation (WHO) (2002) The molecular genetic epidemiology of cystic fibrosis. WHO, Geneva
Acknowledgments
We thank S. Hirtz (CF Centre, University of Heidelberg) for expert technical help with Ussing chamber studies and Dr. D. Seelow (Department of Neuropediatrics, ‘Charité–Universitätsmedizin Berlin’, Berlin, Germany) for critical reading of the manuscript. We also thank Prof. B. Tümmler, Hannover Medical School for inspiring discussion and ideas. Furthermore, we thank Susanne Michel for linguistic proof reading.
This study was supported in part by a grant from the Mukoviszidose e.V. (SIP CFTR-2-2009 to M.A.M).
Conflict of interest
Authors stated that they do not have a financial relationship with the organization that sponsored the research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hentschel, J., Riesener, G., Nelle, H. et al. Homozygous CFTR mutation M348K in a boy with respiratory symptoms and failure to thrive. Disease-causing mutation or benign alteration?. Eur J Pediatr 171, 1039–1046 (2012). https://doi.org/10.1007/s00431-012-1672-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-012-1672-1