
Abstract
Infantile cortical hyperostosis (ICH, OMIM 114000) is a rare familial disorder which affects infants. It spontaneously heals in the first years of life. The disease is characterized by regressive subperiosteal hyperosteogenesis mainly affecting long bones, mandible, clavicles, and ribs which are remarkably swollen and deformed on X-rays. But it is also important to take into consideration the autosomal dominant pattern of inheritance to detect it. In 2005 Gensure et al. detected 3040C→T mutation in COL1A1 gene in three unrelated ICH families. Four generations of patients belonging to the same family were examined in our study. Molecular testing has now disclosed a pathogenic mutation in nine of them. The patients spontaneously recovered. Although our paper shows a distinct correlation between R836C mutation and ICH, there is a certain interindividual and intra-familial variability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Infantile cortical hyperostosis (ICH, OMIM 114000) was first reported by Roske [26] and subsequently studied by De Toni who recognized the congenital and regressive aspects of this disorder [13, 14]. Caffey and Silverman [9] and Caffey [8] formally defined ICH, which is a neonatal and infantile familial disease, spontaneously healing in the first years of life. The disease is characterized by regressive subperiosteal hyperosteogenesis mainly affecting the long bones, mandible, clavicles, and ribs, which are remarkably swollen and deformed on X-ray. The presence of fever, inflammatory signs, and pain led clinicians to suspect different disorders, in particular those caused by infectious agents [2, 22, 25, 30]. Familial clustering has been reported by several authors who also outlined the autosomal dominant pattern of inheritance and a possible reduced penetrance [1, 4, 6, 10, 15, 17, 19, 20, 23, 24, 27, 31].
A genome-wide analysis in three unrelated ICH families allowed Gensure et al. [18] to map the disease locus on chromosome 17q21. All affected individuals were found to be heterozygous for a missense mutation (3040C→T) in the COL1A1 that altered residue 836 (R836C). Suphapeetiporn et al. [29] and Cho et al. [11] corroborated these results by confirming the same mutation in a Thai ICH family and a Korean one.
In the present paper, we report the molecular analysis of the COL1A1 gene in four generations of an ICH Italian family.
Materials and methods
Patients
The affected individuals belong to an Italian Piëmontees family in which ICH has occurred for at least four generations (Figs. 1 and 2). Three of the nine investigated patients were diagnosed in 1969 [15] and one in 1979 [10]. They were reevaluated during this study together with five new patients (IV-11, VI-11, V-3, VII-2, and VII-3). The relevant features of the nine patients are summarized in Table 1.

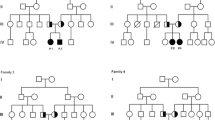
Pedigree of the Italian ICH family. Symbols: arrow, proband; black, affected individuals with COL1A1 mutation; black-gray, heterozygous individual which did not remember ICH data; gray with central black dot, heterozygous asymptomatic; gray, ICH suspected; individuals tested negative are indicated with an horizontal line over their symbol; white, unaffected individuals

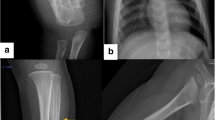
a Patient VI-1 at 14 days old, mandibular deformation. b X-ray of arms. c At 1 year old, bowed arms. d At 3 years old, swelling regression. e At 7 years old, bowed right forearm. f Patient V-5 at 2 months old, swelling of leg. g At 3 years old, full recovery. h Patient VII-2 at 2 months old, X-ray of leg. i, j At 2 years old, light swelling of mandible and leg
In these nine patients and in five asymptomatic family members (VII-1, VI-5, VI-7, VI-9, and VI-10), mutation analysis of COL1A1 was performed. Molecular testing was also performed in a subject (V-1) who was distantly related to her affected husband.
Informed consent was obtained for all patients. Publication of the material included in this work has been authorized in accordance with the terms of the Italian privacy law 196/03.
Methods
DNA extraction and quantification
gDNA was isolated from 300 μl of a peripheral whole-blood sample using PureGene DNA isolation kit according to the manufacturer's instructions (Gentra Systems, Minneapolis, USA). Accurate quantification of obtained gDNA was made by fluorimetric method (Qubit fluorometer; Invitrogen; Carlsbad, USA).
PCR amplification
Amplification of exon 41 of COL1A1 gene was carried out, using specific intronic primers in a 25-μl final volume with 50 ng of gDNA, 250 nM of each primer, 200 μM of each dNTP, 1.5 mM MgCl2, Rxn Buffer, and 1.25 U of Platinum® Taq DNA Polymerase.
A commercial kit (Invitrogen, Carlsbad, USA) was used for Taq, buffer, MgCl2, and dNTPs.
PCR product purification and sequence analysis
The amplified fragment was purified by a selective DNA/silica membrane binding reaction carried out on a column support (Illustra GFX PCR DNA and gel band purification kit; GE Healthcare, Buckinghamshire, UK), and 50 fmol of the same PCR product was used for the dye terminator cycle sequencing reaction with the CEQ DTCS Quick Start Kit (Beckman Coulter, Fullerton, USA) according to the manufacturer's protocol. The sequencing reaction product was purified and analyzed with a capillary electrophoresis sequencer (CEQ 8800; Beckman Coulter, Fullerton, USA). Two different PCR reactions were made and sequenced for each patient to exclude that heterozygotes were due to rare spontaneous polymerization errors of Taq activity.
Results
Mutation analysis of COL1A1 gene (Fig. 3) was performed in 15 subjects. Nine of them (VI-1, VII-2, V-2, VI-4, VII-3, V-5, IV-11, V-3, and VI-11) were found to have a heterozygous Arg836Cys mutation. The onset of the disease began between the first 10 and 60 days of life. Six patients showed evident clinical features of ICH. In particular the following bones were affected: tibial swelling occurred in six patients, ulnar and radial in four, femoral in three, fibular in three, mandibular in two, humeral in two, and clavicular in one (Table 1). For subjects IV-11 and V-3, aged 83 and 71 years respectively, there were no records of any ICH symptoms in the neonatal and infant age although the former is an obligate carrier.

a Representation of COL1A1 gene; the 41 exons are shown as filled boxes and the mutation Arg836Cys related to ICH was also indicated. b Genomic DNA wild type around the site of mutation. c Sequence of a ICH patient with a heterozygous single-nucleotide substitution converting an arginine to cysteine (reference sequence NP_000079.2)
Patient VI-11 is a heterozygous asymptomatic individual. Subjects VII-1, VI-5, VI-7, VI-9, VI-10, and V-1 had a wild-type COL1A1 genotype.
Discussion
The detection of the COL1A1 gene mutation in individuals affected by ICH [18] has shown the molecular make up of this disorder inherited as an autosomal dominant trait, which manifests itself in the first months and years of life and results in early spontaneous recovery. In this study we carried out mutational analysis in 15 subjects of the same family and found heterozygous missense mutation in exon 41 (3040C→T, R836C) in 9 of them.
There have been disagreements on the management of ICH individuals, and the use of corticosteroid therapy has been proposed [7, 12, 16]. However, this therapy was not beneficial in our first patient (VI-1) and after 1 month it was stopped. All the other ICH individuals in the same family were not treated and reached a spontaneous full recovery.
Our study confirms that individuals heterozygous for R836C mutation have a distinct clinical disorder in agreement with the findings of Gensure et al. [18] and Suphapeetiporn et al. [29]. Allelic COL1A1 mutations have been reported in association with Ehlers–Danlos syndrome and osteogenesis imperfecta. None of the known individuals with R836C mutation had clinical or radiographic features consistent with these two diseases (joint hyperlaxity, hyperexstensibility skin, gray-blue sclerae, dentinogenesis imperfecta, premature hearing loss, short stature, deformities, and scoliosis).
There has been some disagreement about the persistence of ICH features in adulthood. A few individuals investigated in the first studies of this disorder were described as having persisting deformities throughout adolescence and adulthood [3, 5, 9]. More recently, Suphapeetiporn et al. [29] have reported two ICH individuals aged 50 and 75 years, with proved COL1A1 mutation, showing persisting deformities of the long bones. The adult mutated individuals studied by Gensure et al. [18] had no skeletal deformation while this was reported for the Thai mutated patients [29]. All our mutated adult individuals reached the target height (Table 1) and at present, they show no persisting ICH symptoms. In four of them, recovery took place between the third and the twelfth years of age. No new X-ray analysis was considered necessary.
Barba and Freriks [1] radiographically reported lesions in the long bones during the last month of gestation in two fetuses that exhibited ICH postnatally and suggested a possible autosomal recessive pattern of inheritance. Other studies have argued that the prenatal onset of ICH should be regarded as a distinct disorder [27]. Schweiger et al. [28] reported 43 cases with prenatal onset of ICH and showed that those born before 35 weeks of gestation had a poor prognosis (possible autosomal recessive inheritance) compared to those born after 35 weeks, who were shown to have an autosomal dominant mutation. It has been suggested that early onset of fetus ICH is usually lethal because of massive hepatic myeloid hyperplasia resulting in enlarged liver, ascites, anasarca, and pulmonary hypoplasia [32]. However, mutational analysis of COL1A1 gene in two fetuses manifesting ICH characteristics disclosed a wild genotype [18]. Kamoun-Goldrat et al. [21] reported an ICH fetus terminated at 30 weeks after a diagnosis of severe osteogenesis imperfecta. A postmortem test confirmed ICH. No patient in our family developed any prenatal ICH symptom.
In conclusion, the mutational origins of this disease are important in making a correct diagnosis and ensuring ICH patients are given the right treatment. Available data point to a wide clinical variability among individuals with R836C mutation. Different outcomes have been observed in a minority of these patients in agreement with the interindividual and intra-familial variability to be expected for an autosomal dominant disorder.
References
Barba WP, Freriks DJ (1953) The familial occurrence of infantile cortical hyperostosis in utero. J Pediatr 42:141
Bernstein RM, Zaleske DJ (1995) Familial aspects of Caffey's disease. Am J Orthop 24:777–781
Blank E (1975) Recurrent Caffey's cortical hyperostosis and persistent deformity. Pediatrics 55:856–860
Boer G, Battistella P (1966) Il fattore genetico nell'iperostosi corticale infantile (sindrome di de Toni-Caffey-Silvermann). Minerva Pediatr 18:1291–1299
Borochowitz Z, Gozal D, Misselevitch I et al (1991) Familial Caffey's disease and late recurrence in a child. Clin Genet 40:329–335
Boyes JG, Demy NG (1951) Infantile cortical hyperostosis: a familial disease? Am J Roentgenol Radium Ther 65:924–930
Bush L, Merrell O (1952) Infantile cortical hyperostosis. Report of a case responding to treatment with corticotrophin. J Pediatr 40:330–333
Caffey J (1957) Infantile cortical hyperostosis; a review of the clinical and radiographic features. Proc R Soc Med 50:347–354
Caffey J, Silverman WA (1945) Infantile cortical hyperostosis: preliminary report on a new syndrome. Am J Roentgenol Radium Ther 54:1
Cerruti Mainardi P (1979) Infantile cortical hyperostosis with raised immunoglobulins. Arch Dis Child 54:985
Cho TJ, Moon HJ, Cho DY et al (2008) The c.3040C>T mutation in COL1A1 is recurrent in Korean patients with infantile cortical hyperostosis (Caffey disease). J Hum Genet 53:947–949
Couper RT, McPhee A, Morris L (2001) Indomethacin treatment of infantile cortical periostosis in twins. J Paediatr Child Health 37:305–308
De Toni G (1943) Una nuova osteopatia infantile: la poliosteopatia deformante connatale regressiva. Policl Infantil No. 6 Anno XI-XII and Radiot. Fis Med 9:1
De Toni G (1953) L'hyperostéogénèse périosto-enchondrale régressive du foetus et du nourrisson. Arch Fr Pédiatr 10:1076–1083
Duillo MT, Cerruti Mainardi P (1969) Sulla familiarità della osteopatia tipo Roske-de Toni-Caffey. Il Lattante 40:706–731
Dutta S, Jain N, Bhattacharya A et al (2005) Infantile cortical hyperostosis. Indian Pediatr 42:64–66
Frana L, Sekanina M (1976) Infantile cortical hyperostosis. Arch Dis Child 51:589–595
Gensure RC, Mäkitie O, Barclay C et al (2005) A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J Clin Invest 115:1250–1257
Gerrard JW, Holman GH, Gorman AA et al (1961) Familial infantile cortical hyperostosis. J Pediatr 59:543–548
Granone FG, Widmer U (1953) Sindrome di De Toni-Caffey. Accad Med 68:431–440
Kamoun-Goldrat A, Martinovic J, Saada J et al (2008) Prenatal cortical hyperostosis with COL1A1 gene mutation. Am J Med Genet A 146A:1820–1824
Kumar TS, Scott JX, Mathew LG (2008) Caffey's disease with raised immunoglobulin levels and thrombocytosis. Indian J Pediatr 75:181–182
Maclachlan AK, Gerrard JW, Houston CS et al (1984) Familial infantile cortical hyperostosis in a large Canadian family. Can Med Assoc J 130:1172–1174
Newberg AH, Tampas JP (1981) Familial infantile cortical hyperostosis: an update. Am J Roentgenol 137:93–96
Ramchander V, Ramkissoon R (1978) Infantile cortical hyperostosis with raised immunoglobulins. Arch Dis Child 53:426–428
Roske G (1930) Eine eigenartige Knochenerkrankung im Säuglingsalter. Mschr Kinderheilk 47:385
Saul RA, Lee WH, Stevenson RE (1982) Caffey's disease revisited. Further evidence for autosomal dominant inheritance with incomplete penetrance. Am J Dis Child 136:55–60
Schweiger S, Chaoui R, Tennstedt C et al (2003) Antenatal onset of cortical hyperostosis (Caffey disease): case report and review. Am J Med Genet A 120A:547–552
Suphapeetiporn K, Tongkobpetch S, Mahayosnond A et al (2007) Expanding the phenotypic spectrum of Caffey disease. Clin Genet 71:280–284
Temperley IJ, Douglas SJ, Rees JP (1972) Raised immunoglobulin levels and thrombocytosis in infantile cortical hyperostosis. Arch Dis Child 47:982–983
Van Buskirk FW, Tampas JP, Peterson OS Jr (1961) Infantile cortical hyperostosis: an inquiry into its familial aspects. Am J Roentgenol Radium Ther Nucl Med 85:613–632
Wright JR Jr, Van den Hof MC, Macken MB (2005) Prenatal infantile cortical hyperostosis (Caffey's disease): a hepatic myeloid hyperplasia-pulmonary hypoplasia sequence can explain the lethality of early onset cases. Prenat Diagn 25:939–944
Acknowledgments
We are grateful to the families for their cooperation and availability. The authors wish to thank Fondazione Cassa di Risparmio di Vercelli for the support and Dr. Michela Godi, Dr. Vanessa Arcuri, and the families for their collaboration.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Cerruti-Mainardi, P., Venturi, G., Spunton, M. et al. Infantile cortical hyperostosis and COL1A1 mutation in four generations. Eur J Pediatr 170, 1385–1390 (2011). https://doi.org/10.1007/s00431-011-1463-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-011-1463-0