
Abstract
Cellular organelles form multiple junctional complexes with one another and the emerging research area dealing with such structures and their functions is undergoing explosive growth. A new research journal named “Contact” has been recently established to facilitate the development of this research field. The current consensus is to define an organellar junction by the maximal distance between the participating organelles; and the gap of 30 nm or less is considered appropriate for classifying such structures as junctions or membrane contact sites. Ideally, the organellar junction should have a functional significance, i.e. facilitate transfer of calcium, sterols, phospholipids, iron and possibly other substances between the organelles (Carrasco and Meyer in Annu Rev Biochem 80:973–1000, 2011; Csordas et al. in Trends Cell Biol 28:523–540, 2018; Phillips and Voeltz in Nat Rev Mol Cell Biol 17:69–82, 2016; Prinz in J Cell Biol 205:759–769, 2014). It is also important to note that the junction is not just a result of a random organelle collision but have active and specific formation, stabilisation and disassembly mechanisms. The nature of these mechanisms and their role in physiology/pathophysiology are the main focus of an emerging research field. In this review, we will briefly describe junctional complexes formed by cellular organelles and then focus on the junctional complexes that are formed by mitochondria with other organelles and the role of these complexes in regulating Ca2+ signalling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Junctions between cellular organelles
The prominent role of junctions between the endoplasmic reticulum (ER) and the plasma membrane (PM) in the regulation of Ca2+ signalling and lipid transport has been recently identified (reviewed in [19, 125]). The discovery that a fundamental signalling process—store operated Ca2+ entry (SOCE) (reviewed in [69, 99, 128, 132]) requires the direct interaction of two relatively small proteins (STIM and Orai) anchored in different organellar membranes (the ER membrane and the PM membrane [45, 101, 105, 142]) attracted considerable interest from cell physiologists and stimulated interest in the formation of the platforms for such interactions, i.e. ER-PM junctions. SOCE is vital for Ca2+ reloading of the ER and for maintaining Ca2+ signalling (reviewed in [128]). Other recently identified, specific functions of Ca2+ signalling micro-domains generated in the ER-PM junctions (e.g. [81]) further highlighted the importance of these signalling platforms.
cAMP signalling is another signalling modality operating in the ER-PM junctions; studies from the A. Hofer laboratory recently defined a novel mechanism of cAMP signalling SOcAMPS (store-operated cAMP signalling) which is activated by ER Ca2+ store depletion and involves the activation of adenylyl cyclase 3 by STIM [97, 107]. Another form of interplay between Ca2+ and cAMP signalling in the ER-PM junctions was extensively characterised in a number of elegant papers by D. Willoughby and colleagues from D. Cooper laboratory. This mechanism involves the direct interaction of adenylyl cyclase 8 and Orai1 [169, 170]. In addition to serving as platforms for SOCE and SOcAMPS, the ER-PM junctions play important roles in the transport of phospholipids (e.g. [21, 25, 152]) and sterols [56, 146].
The molecular mechanism of ER-PM junction formation was first discovered and characterised in yeasts where three groups of proteins Ist2, tricalbin proteins Tcb1–3 and Scs2/Sc22 contribute to the tethering of the organelles ([102, 110] reviewed in [135]). Extended synaptotagmins (mammalian analogues of tricalbins) were later shown to mediate the formation of ER-PM junctions in mammalian cells [22, 60]. The ER is a particularly prominent organelle in its ability to form junctions.
ER junctions with endosomes have been described and are important for the regulation of dynamics and fission of these organelles [51, 143, 174]. It is also likely that the ER-endosomal/lysosomal junctional complexes are important for the coordination of Ca2+ signalling between these organelles [84, 85, 103, 104, 118]). ER junctions with Golgi are essential for the transfer of lipids between the two organelles [114, 115]. Recent study utilising advanced optical spectral microscopy revealed that ER is the preferred interacting organelle for Golgi, peroxisomes and lipid droplets (LDs) in mammalian cells [159].
Membrane tethering between ER and Golgi is mediated by the oxysterol-binding protein (OSBP), which also serves as a conduit for the transfer of sterols and phospholipids between these two organelles [114, 115].
Direct non-vesicular lipid transfer operates between the ER and peroxisomes [138]. Tethers between these organelles have been visualised in the 80s of the previous century [173]. Recently, the proteins responsible for tethering ER and peroxisomes (Pex3p and Inp1p) have been identified in yeasts [88]. This function in mammalian cells is mediated by ACBD5 and VAPB [28].
Interaction between the ER and LDs is important for the lipid transfer to LDs; a complex consisting of fatty acid transport protein 1 (FATP1) and diacylglycerol O-acyltransferase 2 (DGAT2) have been identified as important for both ER-LD interaction and the lipid loading of LDs [172]. Another protein seipin was recently shown to be important for the ER-LD contacts as well as being involved in lipid and protein delivery from ER to LD [144].
ER contacts with phagosomes generate highly localised Ca2+ signals important for phagocytosis [124]. Both junctate and STIM1 are involved in the formation of the junctions between the ER and phagosomes and, interestingly, support different forms of localised Ca2+ responses [62, 124].
Junctions between the ER and other cellular organelles are probably the most numerous inter-organellar junctions. However, junctions formed by other organelles have also been described and include contacts of LDs with peroxisomes and lysosomes (reviewed in [54]), and contacts of lysosomes with peroxisomes [24]. Importantly for the purposes of this review many organelles also form contacts with mitochondria.
Mitochondrial contacts with other cellular organelles
Mitochondria interact and form junctions with LD [6]. Perilipin 5 was shown to be important for this organellar linkage [167]. Another study indicates the importance of mitofusin 2 and perilipin 1 in mediating the interaction between mitochondria and LD [14]. Interestingly, the composition of peridroplet mitochondria and their bioenergetics capacity was shown to be different from their cytoplasmic neighbours [6].
The components of the contact sites between mitochondria and peroxisomes have been characterised using a genome-wide screen in yeast. Pex11 and Mdm34 have been identified as interacting partners involved in the formation of junctions between these cellular organelles [112].
Contacts between mitochondria and Golgi have been described in experimental papers utilising optical microscopy [38, 159]. Interestingly, triple contacts between mitochondria, ER and the Golgi apparatus have been recently identified [159]. Golgi-mitochondrial contacts could be important for Ca2+ signalling in both organelles [38].
Mitochondria-lysosome contacts have also been described in mammalian cells [167, 171]. In another study, mitochondria-lysosomal contacts were systematically investigated using a plethora of microscopy and molecular biology techniques. The observed contacts were tight (approximately 10 nm between the membranes of the participating organelles) and were associated with mitochondrial fission [171]. Two proteins, mitochondrial FISI1 and lysosomal RAB7, were reported to regulate the formation and dissolution of the contacts. Specifically, GTP-bound RAB7 induced the formation of contacts, whilst GDP bound RAB7 dissolved contacts. Conversion from GTP bound to GDP bound forms of RAB7 was facilitated by the GTPase-activating protein TBC1D15, recruited to the contact sites by interaction with FISI1 [171]. Notably, the involvement of a RAB GTPase in the formation of the junctions between the vacuole (lysosome-like structure) and mitochondria was earlier demonstrated in yeasts [70].
Direct contact between endosomes and mitochondria is utilised for the iron transfer from transferrin receptor-containing endosomes to the mitochondria [33, 149]. Interestingly, most of the interactions between these two organelles were short-lived (< 0.5 s), illustrating the notion that organellar junctions do not need to be stable or long-lasting to fulfil physiologically important roles [33].
Mitochondrial junctions with other cellular organelles are schematically illustrated on Fig. 1.

Mitochondrial junctions/interactions with other cellular organelles. Abbreviations in this figure: plasma membrane (PM), endoplasmic reticulum (ER), smooth ER (SER); rough ER (RER); lipid droplet (LD); peroxisome (PRX); endosomes/lysosomes (E/L); Golgi (G). The tethers linking the organelles are indicated by short black bars. Two types of mitochondrial junctions with the SER are included in the figure. The lower mitochondrial-SER junction (SER strand approaches perpendicularly to the mitochondrial outer membrane) illustrates the interaction involved in mitochondrial fission [20, 50, 90]). The upper mitochondrial-SER junction (membranes of the organelles are running parallel to one another) is involved in signalling and lipid transfer between the organelles but not in mitochondrial fission. Note the difference in the length of the tethers between the mitochondrial-SER junctions and mitochondrial-RER junctions (see [29]). A number of triple organellar junctions have been reported (e.g. [159]); in this diagram, we show a putative triple mitochondria-PM-ER junction. Two or three types of tethers could be formed in the triple junctions (two types is the minimal requirement); in this diagram, we show the three types of tethers for illustrative purposes. The strand of ER approaching the PM in the proximity of the ER-PM junction could be SEM [126] or REM [106] but only ribosome-free ER membranes have been shown to form junctions with PM [106, 126]. The properties of the ER and PM in the triple contact regions with mitochondria require further investigations
ER-mitochondria junctions
Early indications of connections between these two organelles have been published in the 1950s of the previous century [9, 26]. Considering the short distance (< 30 nm) between cellular organelles that should be bridged by tethers to form the junctions, electron microscopy (EM) technique is the preeminent methodology in this rapidly developing research field. The contacts between mitochondria and the ER have been indeed visualised by EM (an example is shown on the Fig. 2); furthermore, tethers between the two organelles were also documented in experiments utilising electron tomography [29]. The length of the tethers between strands of smooth ER and mitochondria was approximately 10 nm, whilst the distance bridged by the tethers connecting rough ER and mitochondria was approximately 25 nm [29]. This and other EM studies complemented biochemical observations that a specific fraction of the ER is associated with mitochondria. This fraction is termed MAM (mitochondria-associated membranes). It is important for phospholipid synthesis and the transport of phospholipids between the ER and mitochondria, including the transfer of phosphatidylserine from the ER to mitochondria and of phosphatidylethanolamine from mitochondria to the ER (early evidence [161], recent reviews [98, 132, 162]). The specific biochemical procedures involved in the isolation of MAMs are described in a recent review by J. Vance [162]. Importantly, in most cell types, MAMs are identified/characterised by proteins that are not unique in MAMs but are enriched in MAMs [162]. These proteins include phosphatidylserine synthase-1 and synthase-2 [153], Sigma-1 receptor [65], Mitofusin 2 [34] and, importantly for this review, inositol trisphosphate receptors (IP3R) [140, 154].

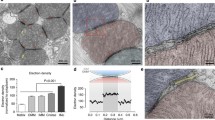
Mitochondria can be found in close proximity to the endoplasmic reticulum and the plasma membrane in pancreatic acinar cells. a Images of mitochondria in live pancreatic acinar cells (adapted with modifications from [165]). Mitochondria were loaded with the ΔΨ indicator TMRM (tetramethylrhodamine methyl ester). SP-M indicates subplasmalemmal mitochondria (see also parts c and d of this figure and [76, 129]). PG-M indicates perigranular mitochondria. In this cell type, PG-M can be found in close proximity to Golgi, ER strands and secretory granules (see [38, 76, 129, 158]). Scale bar corresponds to 4 μm. b Example a mitochondrion located in a close proximity to a rough ER strand (adapted with modifications from [76]). Ri indicates ribosome. ER-L indicates the ER lumen. Scale bar represents 100 nm. c ER-PM junctions (indicated by arrowheads) with associated mitochondrion (m). The image is adapted with modifications from [106]). The lumens of ER strands approaching the plasma membranes are highlighted by asterisks. This image is an example of a triple organellar junction in a primary mammalian cell. Scale bar represents approximately 50 nm. d Subplasmalemmal mitochondrion (SP-M) shown with associated plasma membrane (PM) region. Note the strands of the rough ER in close proximity to the mitochondrion on the other side from the PM. Scale bar corresponds to approximately 100 nm. The figure was adapted with modifications from [76]
Recently, there was considerable progress in the characterisation of the molecular composition of the tethers linking ER with the outer mitochondrial membrane (OMM). In yeasts, a complex termed ERMES (ER-mitochondria encounter structure) has been identified by exceptionally elegant experiments combining expression of artificial tether linking ER with mitochondria and analysis of mutations in yeasts colonies. These experiments identified components of ERMES on the basis that mutations or deletions of these components results in growth deficiency that could be rescued by the expression of the artificial tether [89]. The identified in these experiments proteins Mmm1/Mdm10/Mdm12/Mdm34 form ERMES. Two proteins forming ERMES (Mdm34 and Mdm10) are anchored in OMM; whereas Mmm1 is an ER membrane resident and Mdm12 is a cytosolic protein recruited to ERMES complex ([89] reviewed in [95]). A recent study by S. Kawano and colleagues reported that the Mmm1-Mdm12 complex is sufficient for the transfer of phospholipids between membranes [82]. Notably, in yeasts, there is a redundancy of both lipid ER-mitochondria tethering and lipid transfer mechanisms; in particular, conserved EMC (endoplasmic reticulum membrane protein complex) has been identified and suggested to mediate both tethering and lipid transfer functions [94] (reviewed in [119]). Another redundancy is based on the functional substitution of ER-mitochondrial junctions in ERMES impaired yeasts with junctions formed between vacuole and mitochondria termed vCLAMP (vacuole-mitochondria contact patches) [44, 70]). Impairment of ERMES results in the expansion of vCLAMP and vice versa, whereas the elimination of both structures is lethal. Vps39 was shown to be important for vCLAMP formation [44, 70]. Recent studies from the B. Kornmann laboratory indicated that endosomal protein Vps13 and mitochondrial protein Mcp1 mediate the functions of vCLAMP [75, 96].
ERMES complex/components are not retained in metazoans and other types of proteins are responsible for the formation of ER-mitochondria junctions in animal cells. Linkers formed by OMM protein VDAC1 (voltage-dependent anion channel 1), IP3R (located in the ER) and a molecular chaperone glucose-regulated protein 75 (grp75) was suggested by G. Szabadkai and colleagues from R. Rizzutto laboratory [154]. The presence of IP3Rs was later observed in proximity-labelling assays designed to reveal the proteome of ER-mitochondrial junctions [23]. The composition of such linker is clearly beneficial for the Ca2+ transfer from the ER to mitochondria. Mitofusin 2 was suggested as the linker between the ER and mitochondria [34]. This notion has been challenged (e.g. [27, 46]) and the debate currently continues [47, 48, 121, 122]. Recently, a number of ER proteins interacting with mitochondrial proteins and therefore capable, in principle, to serve as tethers have been reported; they include: vesicle-associated membrane protein-associated protein B (VAPB) interacting with mitochondrial protein tyrosine phosphatase interacting protein 51 (PTPIP51) [36]; oxysterol-binding proteins (OSBP)-related proteins ORP5 and ORP8 that also interact with PTPIP51 [52]; Bap31 that interact with mitochondria-located Fission 1 [174] and ribosome-binding protein 1 (RRBP1) with its mitochondrial binding partner Synaptojanin-2-binding protein (SYNJ2BP) [73].
Among the regulators of ER-mitochondrial junctions are ER-shaping proteins reticulons, which were identified as a result of ascorbate peroxidase proximity labelling [23]. The ability of reticulons (specifically of RTN1A, RTN2B, and RTN3B) to increase ER-mitochondria interaction was determined by split luciferase assay [23]. Similar technique was used to ascertain the role of receptor expression-enhancing protein 1 (REEP1) in potentiating ER-mitochondria interaction [100]. Another regulator of ER-mitochondrial junctions is the endoplasmic-reticulum-associated E3 ubiquitin ligase Gp78, which is particularly important for rough ER-mitochondria contacts [168].
Mitochondrial fission requires the formation of specific contacts between mitochondrial membrane and the ER. Dynamin related protein 1 (Drp1) is essential for mitochondrial fission (e.g. [92, 151] reviewed in [160]). A study by J. Friedman and colleagues from the G. Voeltz laboratory reported that the location of mitochondrial division is determined by contact with the ER tubule, which is formed before the recruitment of Drp1 to the inter-organellar contact and induces mitochondria constriction at the contact region [50]. The ER in the junction is enriched with inverted formin 2 (INF2) which induces actin filament formation in the junction [90]. Notably, INF2-induced actin recruitment is important for the formation of contacts between the ER and mitochondria [20]
ER-mitochondria junctions as signalling nanodomains
Ca2+ signalling is an important signalling modality operating in the ER-mitochondria junctions. Many biophysical properties of mitochondrial Ca2+ influx and extrusion have been characterised in the second part of the twentieth century. In particular it was established that the mitochondrial Ca2+ influx system operates as “uniporter” (i.e. does not involve accompanying transfer of other ions for charge compensation) and that it can be efficiently inhibited by Ruthenium Red (RuRed). The mitochondrial Ca2+ export system was characterised as Na+/Ca2+ exchanger, which can also transport Ca2+ when Na + is substituted by Li + and can be inhibited by CGP-37157. These crucially important early discoveries are reviewed in [18]. Development of mitochondria specific bioluminescent probes by R. Rizzuto, T. Pozzan and their colleagues has given considerable impetus to the advancement of this research field [139]. Important studies defining the physiological and pathophysiological role of mitochondrial Ca2+ have also been conducted in the last three decades of the twentieth century. R. Denton’s group defined an important role of mitochondrial Ca2+ in the regulation of the Krebs cycle (reviewed in [37, 113]). At the cellular level, changes in the activity of the Krebs cycle can be visualised by recording NAD(P)H and FAD fluorescence (reviewed in [42]). Clear correlation between cytosolic Ca2+, mitochondrial Ca2+ and NADH responses has been indeed recorded (e.g. [63, 164]. The upregulation of Krebs cycle and other Ca2+-dependent mitochondrial reactions underpins the regulation of mitochondrial ATP production required for efficient stimulus-metabolism coupling (e.g. [77, 157, 166], reviewed in [55, 156]).
The role of Ca2+ microdomains and the importance of the contacts between the ER and mitochondria for mitochondrial Ca2+ influx was emphasised by studies from the T. Pozzan laboratory [140, 141]. The importance of the microdomains and organellar contacts was attributed to the relatively low affinity of the mitochondrial uniporter to the cytosolic Ca2+ [140, 141]. The recent rapid development of this research area provided mechanistic explanation to this phenomenon. The direct electrophysiological recordings of the MCU current were reported by Y. Kirichock and colleagues from the D. Clapham laboratory [86]. In 2011, two laboratories independently identified the protein mediating mitochondrial Ca2+ entry and termed it mitochondrial calcium uniporter (MCU) [4, 35]. Approximately 1 year earlier, F. Perocchi and colleagues from the V. Mootha laboratory discovered an important regulator of mitochondrial Ca2+ import, MICU1 [131]. This research area undergone rapid development in the next few years and a number of other regulators of MCU have been discovered including MICU2 [134] and EMRE [145]. Both MICU1 and MCU2 are EF hand-containing Ca2+-binding proteins [131, 134]. An important role of MICU1 and MICU2 in the MCU complex is to form and regulate the threshold of cytosolic Ca2+, which allows efficient Ca2+ entry into the mitochondria (e.g. [31, 78, 80, 109, 130] for review see [79] and the paper by C. Mammucari and colleagues in the current issue). A resting mitochondrial membrane potential (ΔΨ) of approximately − 160 mV is sufficient to drive Ca2+ entry into the mitochondria even at low resting cytosolic Ca2+ concentrations. Increased Ca2+ threshold for the mitochondrial Ca2+ entry is beneficial for the cell since it prevents or reduces Ca2+ entry into the mitochondria at low (resting or near-resting) cytosolic Ca2+ levels. This prevents futile Ca2+ cycle and the associated bioenergetics costs required to maintain acceptably low mitochondrial Ca2+ concentration. Such futile Ca2+ cycle and ATP expenditure were recently demonstrated in cells harbouring MICU1 mutation by G. Bhosale and colleagues from M. Duchen’s laboratory [10]. Threshold created by MICU1 and MICU2 is an important mechanism for reducing the signal-to-noise ratio for the communication between Ca2+ signalling and mitochondria. Importantly, it works in conjunction with Ca2+ signalling microdomains formed in the ER-mitochondrial junctions, which further increase the difference between bulk cytosolic Ca2+ rise and the Ca2+ rise in the proximity to the Ca2+-releasing channels and OMM region located in the junctional complex. Direct measurements of Ca2+ increases in the ER-Mitochondrial junctions have been conducted by G. Csordas and colleagues from the G. Hajnoczky laboratory by placing Ca2+ indicators into the junctions [30]. This study reported high amplitude IP3-induced Ca2+ responses (> 9 μM) in the junctions (substantially higher than the bulk cytosolic Ca2+ increase) and the relative insensitivity of the junctional Ca2+ transients to slow Ca2+ buffering by EGTA [30]. The substantial difference between local Ca2+ signals in the junction and the rest of the cytosol enhances the signal-to-noise ratio for mitochondrial transfer of Ca2+ signals and facilitates this form of stimulus—metabolism coupling. The findings reported by G. Csordas and colleagues were consistent with results reported by M. Giacomello and colleagues who targeted Ca2+ indicator to the OMM and reported the appearance of Ca2+ hot spots where the Ca2+ concentration was found to be more than 5 times higher than that of the bulk cytosolic concentration [57]. The presence of IP3Rs in MAMs and their suggested role as a component of the junctional complex [154] are also in agreement with these findings.
RyRs form another group of intracellular Ca2+-releasing channels particularly prominent in the sarcoplasmic reticulum (a specialised form of the endoplasmic reticulum present in muscle cells). There is now a sufficient body of evidence supporting the formation of SR-mitochondrial junctions and privileged local Ca2+ transfer from RyR into the mitochondria. Electron microscopy imaging revealed close contacts between mitochondrial and SR membranes (e.g. [66]). High Ca2+ concentration hot-spots (> 20 μM) have been recorded on the OMM of cardiomyocytes [39]. Mitochondrial Ca2+ increase following RyRs activation occurs in the presence of cytosolic calcium buffer in cardiac [148, 155] and skeletal [150] muscle cells, confirming the existence of functionally coupled organellar junctions. The Ca2+ transfer by this mechanism is therefore important for stimulus-metabolism coupling in muscle cells ([16, 155] reviewed in [43]).
Mitochondrial Ca2+ transfer in the junctional complexes is important not only for the stimulus-metabolism coupling. A recent study by R. Chakrabarti and colleagues highlighted the importance of Ca2+ influx in ER-mitochondrial junction and Ca2+ entry into the mitochondria via MCU for mitochondrial fission [20].
Mitochondrial Ca2+ is important for the opening of the mitochondrial permeability transition pore (MPTP). MPTP is a high conductance mitochondrial channel permeable to molecules with molecular weight up to 1.5 kDa [40]. The exact role of mitochondrial Ca2+ as permissive or initiating factor in physiological/pathophysiological settings involving MPTP is debated (see [8]). Permissive or inducing, the mitochondrial Ca2+ is important for MPTP opening and therefore for the associated cell/tissue damage. Considering the importance of MPTP in pathophysiology of cardiovascular system (reviewed in [64]) and nervous system (reviewed in [41]), and the significance of ER-Mitochondrial junctional complexes for mitochondrial Ca2+ transfer, one can expect that the role of junctional complexes in pathophysiological conditions will gain considerable attention in the next few years. This process has already began: e.g. a study by L. Hedskog and colleagues suggested the link between the increase in the number of the ER-mitochondrial contacts and the pathophysiology of Alzheimer disease [67], whilst X. Qiao and colleagues highlighted the importance of PTPIP51 (protein regulating ER-mitochondria junction) for ischemia/reperfusion injury [136]. It is safe to predict that the study of the structure, dynamics and role of junctional complexes in diseases will be an important subfield in modern biomedical research.
ER-mitochondrial junctions are also sites of localised H2O2 nanodomains that were recently directly measured and reported by D. Booth and colleagues [12]. In this elegant study from the G. Hajnoczky laboratory, the authors targeted the H2O2 sensor HyPer [5] to the inducible linkers between the ER and mitochondria, and observed Ca2+-dependent redox nanodomains in the junctions between the organelles [12]. Interestingly, H2O2 transients potentiated ER Ca2+ release [12]. Redox regulation of IP3Rs is well documented (e.g. [2, 13] reviewed in [1]) and junctional complexes involving the ER with a ROS producing organelle (i.e. mitochondrion) is prime location for such regulation. Importantly, RyR are also redox sensitive channels (reviewed in [68]) and important sensitivity adjustment of this channel could take place in SR-mitochondrial junctions by locally produced ROS.
Mitochondria-PM junctions and Ca2+ signalling
The mechanisms tethering mitochondria to the plasma membrane have been characterised in yeasts, where the Num1/Mdm36 anchors ER-mitochondria complex to the plasma membrane [87, 93, 133]. Subplasmalemmal mitochondrial groups have been reported in a number of mammalian cell types (e.g. [49, 76, 129, 137] see also Fig. 2) but the mechanism involved in the formation of tethers between the mitochondria and the plasma membrane in mammalian cells is currently unknown.
Using Ca2+ indicators targeted to OMM and the cytosol, Giacomello and colleagues established that mitochondria adjacent to the plasma membrane did not show preferential Ca2+ uptake upon activation of store operated Ca2+ entry [57]. Furthermore, Ca2+ entry via SOCE was ineffective in producing Ca2+ hot spots on the OMM. Nevertheless, Ca2+ entry into the mitochondria was recorded in these experiments and the peak mitochondrial Ca2+ concentration was approximately one order of magnitude higher than in the cytosol [57]. The absence of privilege communication between STIM–Orai channels and mitochondria was also observed in COS-7 cells by M. Korzeniowski and colleagues from the A. Spat laboratory [91].
On the other hand, a study by P. Varadi and colleagues demonstrated that the re-localisation of mitochondria from the plasma membrane results in a clearly resolvable decrease of store operated Ca2+ entry and reduction in mitochondrial Ca2+ responses [163]. This is consistent with the findings by A. Quintana and colleagues from the M. Hoth laboratory which revealed the prominent role of mitochondria in the immunological synapse. In this highly specialised signalling region, essential for T cell activation, mitochondria regulate store operated Ca2+ entry [137]. Importantly, this is achieved by a specialised group of subplasmalemmal mitochondria. The authors concluded that the local subplasmalemmal mitochondria prevent calcium-dependent inactivation of ORAI channels in the immunological synapse and therefore extend/amplify Ca2+ responses. This is achieved as a result of a local coordination of STIM/Orai channels, mitochondria and Ca2+ pumps of the plasma membrane. This study extends previous findings of the importance of mitochondria in the regulation of SOCE and its electrophysiological manifestation Ca2+ release-activated Ca2+ (CRAC) current (ICRAC) ([3, 53, 58, 59, 61, 71, 72, 108, 111] and specifically about the role of mitochondrial Ca2+ buffering in the regulation of ICRAC inactivation [127]). It is conceivable that mitochondria could regulate SOCE/ICRAC not only via local Ca2+ uptake but also by releasing products of mitochondrial metabolism in the proximity to the Ca2+ channel. In particular, it was shown that ATP released from subplasmalemmal mitochondria can facilitate SOCE by providing local Ca2+ buffering [116]. Notably, subplasmalemmal ATP microdomains have been recorded [83]. The authors of this study also suggested that a specific peripheral group of mitochondria is responsible for such micro domains.
Interestingly, the T.Pozzan group demonstrated privilege communication between mitochondria and voltage-gated Ca2+ channels. This study reported that the subplasmalemmal mitochondria are exposed to higher Ca2+ concentrations and show stronger Ca2+ responses than mitochondria located in the deeper regions of the cytoplasm [57]. Similar conclusion was reached in the study by Montero and colleagues that demonstrated very strong Ca2+ increases (hundreds of μM) in a subgroup of mitochondria upon activation of voltage-gated Ca2+ channels [117]. Interestingly, this study suggests a triple functional interaction between voltage-gated Ca2+ channels, RyR and mitochondria [117]. A recent study by A. Valm and colleagues, utilising high-resolution optical microscopy, identified a number of close contacts formed by ERMCSs (ER mitochondria contact sites) with other organelles [159]. The structure-function relationships of such triple organellar junctions will probably form an exciting avenue for further development in this research subfield.
Subplasmalemmal mitochondria regulate not only Ca2+ channels but also Ca2+ pumps. This coordinated regulation is probably needed to ensure balance between Ca2+ signalling and Ca2+ homeostasis. The important role of subplasmalemmal mitochondrial group in the regulation of both SOCE- and PMCA-mediated Ca2+ fluxes was reported M. Frieden and colleagues [49].
Mitochondria are an important source of reactive oxygen species (reviewed in [15, 120, 147]). Both Ca2+ extrusion by PMCA and Ca2+ entry via STIM/Orai channels are redox sensitive processes (e.g. ([11, 17], see also [123]). Recently, mitochondrial ROS was implicated in the regulation of SOCE [7]). Subplasmalemmal mitochondria would be particularly suitable organelles for this form of regulation.
Concluding remarks
One can observe clear indications of the emergence of a new research field focused on the mechanisms contributing to the formation of junctions between cellular organelles and determining functions of the interorganellar complexes. The development of this field is facilitated by the rapid advances in super-resolution microscopy and correlative optical-electron microscopy. This emerging field has already facilitated the development of new molecular biology techniques (e.g. introduction of artificial tethers/linkers that can bridge cellular organelles and can be decorated with sensors of signalling molecules). Development of techniques for selective labelling of junctional proteins and consequently identification of junctional proteomes should provide further impetus to this research area. It is likely that ER-mitochondrial junctions and mitochondria-PM junctions serve as important elements in stimulus-metabolism coupling and that this and other physiological functions of the junctional complexes will be actively investigated in the near future. Understanding the mechanisms involved in the formation and functioning of junctional complexes (and particularly of mitochondrial junctions with other cellular organelles) will be beneficial for elucidating the pathophysiological implications of the disruption of these important transport/signalling platforms.
References
Appenzeller-Herzog C, Simmen T (2016) ER-luminal thiol/selenol-mediated regulation of Ca2+ signalling. Biochem Soc Trans 44:452–459
Bansaghi S, Golenar T, Madesh M, Csordas G, RamachandraRao S, Sharma K, Yule DI, Joseph SK, Hajnoczky G (2014) Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J Biol Chem 289:8170–8181
Barrow SL, Voronina SG, da Silva Xavier G, Chvanov MA, Longbottom RE, Gerasimenko OV, Petersen OH, Rutter GA, Tepikin AV (2008) ATP depletion inhibits Ca2+ release, influx and extrusion in pancreatic acinar cells but not pathological Ca2+ responses induced by bile. Pflugers Arch 455:1025–1039
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476:341–345
Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, Terskikh AV, Lukyanov S (2006) Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat Methods 3:281–286
Benador IY, Veliova M, Mahdaviani K, Petcherski A, Wikstrom JD, Assali EA, Acin-Perez R, Shum M, Oliveira MF, Cinti S, Sztalryd C, Barshop WD, Wohlschlegel JA, Corkey BE, Liesa M, Shirihai OS. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics (2018) Composition, and dynamics that support lipid droplet expansion. Cell Metab 27:869–885 e866
Ben-Kasus Nissim T, Zhang X, Elazar A, Roy S, Stolwijk JA, Zhou Y, Motiani RK, Gueguinou M, Hempel N, Hershfinkel M, Gill DL, Trebak M, Sekler I (2017) Mitochondria control store-operated ca(2+) entry through Na(+) and redox signals. EMBO J 36:797–815
Bernardi P, Di Lisa F (2015) The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 78:100–106
Bernhard W, Rouiller C (1956) Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J Biophys Biochem Cytol 2:73–78
Bhosale G, Sharpe JA, Koh A, Kouli A, Szabadkai G, Duchen MR (2017) Pathological consequences of MICU1 mutations on mitochondrial calcium signalling and bioenergetics. Biochim Biophys Acta 1864:1009–1017
Bogeski I, Kummerow C, Al-Ansary D, Schwarz EC, Koehler R, Kozai D, Takahashi N, Peinelt C, Griesemer D, Bozem M, Mori Y, Hoth M, Niemeyer BA (2010) Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal 3:ra24
Booth DM, Enyedi B, Geiszt M, Varnai P, Hajnoczky G (2016) Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol Cell 63:240–248
Bootman MD, Taylor CW, Berridge MJ (1992) The thiol reagent, thimerosal, evokes Ca2+ spikes in HeLa cells by sensitizing the inositol 1,4,5-trisphosphate receptor. J Biol Chem 267:25113–25119
Boutant M, Kulkarni SS, Joffraud M, Ratajczak J, Valera-Alberni M, Combe R, Zorzano A, Canto C (2017) Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J 36:1543–1558
Brand MD (2010) The sites and topology of mitochondrial superoxide production. Exp Gerontol 45:466–472
Brandes R, Bers DM (1997) Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ Res 80:82–87
Bruce JI, Elliott AC (2007) Oxidant-impaired intracellular Ca2+ signaling in pancreatic acinar cells: role of the plasma membrane Ca2+-ATPase. Am J Physiol Cell Physiol 293:C938–C950
Carafoli E (2012) The interplay of mitochondria with calcium: an historical appraisal. Cell Calcium 52:1–8
Carrasco S, Meyer T (2011) STIM proteins and the endoplasmic reticulum-plasma membrane junctions. Annu Rev Biochem 80:973–1000
Chakrabarti R, Ji WK, Stan RV, de Juan Sanz J, Ryan TA, Higgs HN (2018) INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J Cell Biol 217:251–268
Chang CL, Liou J (2015) Phosphatidylinositol 4,5-bisphosphate homeostasis regulated by Nir2 and Nir3 proteins at endoplasmic reticulum-plasma membrane junctions. J Biol Chem 290:14289–14301
Chang CL, Hsieh TS, Yang TT, Rothberg KG, Azizoglu DB, Volk E, Liao JC, Liou J (2013) Feedback regulation of receptor-induced Ca2+ signaling mediated by E-Syt1 and Nir2 at endoplasmic reticulum-plasma membrane junctions. Cell Rep 5:813–825
Cho IT, Adelmant G, Lim Y, Marto JA, Cho G, Golden JA (2017) Ascorbate peroxidase proximity labeling coupled with biochemical fractionation identifies promoters of endoplasmic reticulum-mitochondrial contacts. J Biol Chem 292:16382–16392
Chu BB, Liao YC, Qi W, Xie C, Du X, Wang J, Yang H, Miao HH, Li BL, Song BL (2015) Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 161:291–306
Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, De Camilli P, INTRACELLULAR TRANSPORT (2015) PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 349:428–432
Copeland DE, Dalton AJ (1959) An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J Biophys Biochem Cytol 5:393–396
Cosson P, Marchetti A, Ravazzola M, Orci L (2012) Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PLoS One 7:e46293
Costello JL, Castro IG, Hacker C, Schrader TA, Metz J, Zeuschner D, Azadi AS, Godinho LF, Costina V, Findeisen P, Manner A, Islinger M, Schrader M (2017) ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J Cell Biol 216:331–342
Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174:915–921
Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, Balla T, Hajnoczky G (2010) Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell 39:121–132
Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnoczky G (2013) MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab 17:976–987
Csordas G, Weaver D, Hajnoczky G (2018) Endoplasmic reticular-mitochondrial contactology: structure and signaling functions. Trends Cell Biol 28:523–540
Das A, Nag S, Mason AB, Barroso MM (2016) Endosome-mitochondria interactions are modulated by iron release from transferrin. J Cell Biol 214:831–845
de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456:605–610
De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476:336–340
De Vos KJ, Morotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, Warley A, Shaw CE, Miller CC (2012) VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet 21:1299–1311
Denton RM (2009) Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787:1309–1316
Dolman NJ, Gerasimenko JV, Gerasimenko OV, Voronina SG, Petersen OH, Tepikin AV (2005) Stable Golgi-mitochondria complexes and formation of Golgi Ca(2+) gradients in pancreatic acinar cells. J Biol Chem 280:15794–15799
Drago I, De Stefani D, Rizzuto R, Pozzan T (2012) Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci U S A 109:12986–12991
Duchen MR (2000) Mitochondria and Ca(2+)in cell physiology and pathophysiology. Cell Calcium 28:339–348
Duchen MR (2012) Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch 464:111–121
Duchen MR, Surin A, Jacobson J (2003) Imaging mitochondrial function in intact cells. Methods Enzymol 361:353–389
Eisner V, Csordas G, Hajnoczky G (2013) Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca(2)(+) and reactive oxygen species signaling. J Cell Sci 126:2965–2978
Elbaz-Alon Y, Rosenfeld-Gur E, Shinder V, Futerman AH, Geiger T, Schuldiner M (2014) A dynamic interface between vacuoles and mitochondria in yeast. Dev Cell 30:95–102
Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441:179–185
Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P (2015) Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci U S A 112:E2174–E2181
Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P (2016) Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell Rep 15:2226–2238
Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P (2017) On the role of Mitofusin 2 in endoplasmic reticulum-mitochondria tethering. Proc Natl Acad Sci U S A 114:E2266–E2267
Frieden M, Arnaudeau S, Castelbou C, Demaurex N (2005) Subplasmalemmal mitochondria modulate the activity of plasma membrane Ca2+-ATPases. J Biol Chem 280:43198–43208
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334:358–362
Friedman JR, Dibenedetto JR, West M, Rowland AA, Voeltz GK (2013) Endoplasmic reticulum-endosome contact increases as endosomes traffic and mature. Mol Biol Cell 24:1030–1040
Galmes R, Houcine A, van Vliet AR, Agostinis P, Jackson CL, Giordano F (2016) ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep 17:800–810
Gamberucci A, Innocenti B, Fulceri R, Banhegyi G, Giunti R, Pozzan T, Benedetti A (1994) Modulation of Ca2+ influx dependent on store depletion by intracellular adenine-guanine nucleotide levels. J Biol Chem 269:23597–23602
Gao Q, Goodman JM (2015) The lipid droplet-a well-connected organelle. Front Cell Dev Biol 3:49
Gaspers LD, Memin E, Thomas AP (2012) Calcium-dependent physiologic and pathologic stimulus-metabolic response coupling in hepatocytes. Cell Calcium 52:93–102
Gatta AT, Wong LH, Sere YY, Calderon-Norena DM, Cockcroft S, Menon AK, Levine TP (2015) A new family of StART domain proteins at membrane contact sites has a role in ER-PM sterol transport. eLife 4:e07253, https://doi.org/10.7554/eLife.07253
Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T (2010) Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell 38:280–290
Gilabert JA, Parekh AB (2000) Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca(2+) current I(CRAC). EMBO J 19:6401–6407
Gilabert JA, Bakowski D, Parekh AB (2001) Energized mitochondria increase the dynamic range over which inositol 1,4,5-trisphosphate activates store-operated calcium influx. EMBO J 20:2672–2679
Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva EO, Bagriantsev SN, Borgese N, De Camilli P (2013) PI(4,5)P(2)-dependent and Ca(2+)-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell 153:1494–1509
Glitsch MD, Bakowski D, Parekh AB (2002) Store-operated Ca2+ entry depends on mitochondrial Ca2+ uptake. EMBO J 21:6744–6754
Guido D, Demaurex N, Nunes P (2015) Junctate boosts phagocytosis by recruiting endoplasmic reticulum Ca2+ stores near phagosomes. J Cell Sci 128:4074–4082
Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82:415–424
Halestrap AP, Pasdois P (2009) The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta 1787:1402–1415
Hayashi T, Su TP (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate ca(2+) signaling and cell survival. Cell 131:596–610
Hayashi T, Martone ME, Yu Z, Thor A, Doi M, Holst MJ, Ellisman MH, Hoshijima M (2009) Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Sci 122:1005–1013
Hedskog L, Pinho CM, Filadi R, Ronnback A, Hertwig L, Wiehager B, Larssen P, Gellhaar S, Sandebring A, Westerlund M, Graff C, Winblad B, Galter D, Behbahani H, Pizzo P, Glaser E, Ankarcrona M (2013) Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer's disease and related models. Proc Natl Acad Sci U S A 110:7916–7921
Hidalgo C, Donoso P, Carrasco MA (2005) The ryanodine receptors Ca2+ release channels: cellular redox sensors? IUBMB Life 57:315–322
Hogan PG, Rao A (2015) Store-operated calcium entry: mechanisms and modulation. Biochem Biophys Res Commun 460:40–49
Honscher C, Mari M, Auffarth K, Bohnert M, Griffith J, Geerts W, van der Laan M, Cabrera M, Reggiori F, Ungermann C (2014) Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev Cell 30:86–94
Hoth M, Fanger CM, Lewis RS (1997) Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol 137:633–648
Hoth M, Button DC, Lewis RS (2000) Mitochondrial control of calcium-channel gating: a mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proc Natl Acad Sci U S A 97:10607–10612
Hung V, Lam SS, Udeshi ND, Svinkina T, Guzman G, Mootha VK, Carr SA, Ting AY (2017) Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 6
Iwasawa R, Mahul-Mellier AL, Datler C, Pazarentzos E, Grimm S (2011) Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J 30:556–568
John Peter AT, Herrmann B, Antunes D, Rapaport D, Dimmer KS, Kornmann B (2017) Vps13-Mcp1 interact at vacuole-mitochondria interfaces and bypass ER-mitochondria contact sites. J Cell Biol 216:3219–3229
Johnson PR, Dolman NJ, Pope M, Vaillant C, Petersen OH, Tepikin AV, Erdemli G (2003) Non-uniform distribution of mitochondria in pancreatic acinar cells. Cell Tissue Res 313:37–45
Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R (1999) Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A 96:13807–13812
Kamer KJ, Mootha VK (2014) MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep 15:299–307
Kamer KJ, Mootha VK (2015) The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol 16:545–553
Kamer KJ, Grabarek Z, Mootha VK (2017) High-affinity cooperative ca(2+) binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep 18:1397–1411
Kar P, Nelson C, Parekh AB (2011) Selective activation of the transcription factor NFAT1 by calcium microdomains near Ca2+ release-activated Ca2+ (CRAC) channels. J Biol Chem 286:14795–14803
Kawano S, Tamura Y, Kojima R, Bala S, Asai E, Michel AH, Kornmann B, Riezman I, Riezman H, Sakae Y, Okamoto Y, Endo T (2018) Structure-function insights into direct lipid transfer between membranes by Mmm1-Mdm12 of ERMES. J Cell Biol 217:959–974
Kennedy HJ, Pouli AE, Ainscow EK, Jouaville LS, Rizzuto R, Rutter GA (1999) Glucose generates sub-plasma membrane ATP microdomains in single islet beta-cells. Potential role for strategically located mitochondria. J Biol Chem 274:13281–13291
Kilpatrick BS, Eden ER, Schapira AH, Futter CE, Patel S (2013) Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J Cell Sci 126:60–66
Kilpatrick BS, Eden ER, Hockey LN, Yates E, Futter CE, Patel S (2017) An endosomal NAADP-sensitive two-pore Ca(2+) channel regulates ER-endosome membrane contact sites to control growth factor signaling. Cell Rep 18:1636–1645
Kirichok Y, Krapivinsky G, Clapham DE (2004) The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427:360–364
Klecker T, Scholz D, Fortsch J, Westermann B (2013) The yeast cell cortical protein Num1 integrates mitochondrial dynamics into cellular architecture. J Cell Sci 126:2924–2930
Knoblach B, Sun X, Coquelle N, Fagarasanu A, Poirier RL, Rachubinski RA (2013) An ER-peroxisome tether exerts peroxisome population control in yeast. EMBO J 32:2439–2453
Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P (2009) An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325:477–481
Korobova F, Ramabhadran V, Higgs HN (2013) An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339:464–467
Korzeniowski MK, Szanda G, Balla T, Spat A (2009) Store-operated Ca2+ influx and subplasmalemmal mitochondria. Cell Calcium 46:49–55
Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM (1999) C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell 4:815–826
Lackner LL, Ping H, Graef M, Murley A, Nunnari J (2013) Endoplasmic reticulum-associated mitochondria-cortex tether functions in the distribution and inheritance of mitochondria. Proc Natl Acad Sci U S A 110:E458–E467
Lahiri S, Chao JT, Tavassoli S, Wong AK, Choudhary V, Young BP, Loewen CJ, Prinz WA (2014) A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol 12:e1001969
Lang A, John Peter AT, Kornmann B (2015a) ER-mitochondria contact sites in yeast: beyond the myths of ERMES. Curr Opin Cell Biol 35:7–12
Lang AB, John Peter AT, Walter P, Kornmann B (2015b) ER-mitochondrial junctions can be bypassed by dominant mutations in the endosomal protein Vps13. J Cell Biol 210:883–890
Lefkimmiatis K, Srikanthan M, Maiellaro I, Moyer MP, Curci S, Hofer AM (2009) Store-operated cyclic AMP signalling mediated by STIM1. Nat Cell Biol 11:433–442
Lev S (2010) Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat Rev Mol Cell Biol 11:739–750
Lewis RS (2007) The molecular choreography of a store-operated calcium channel. Nature 446:284–287
Lim Y, Cho IT, Schoel LJ, Cho G, Golden JA (2015) Hereditary spastic paraplegia-linked REEP1 modulates endoplasmic reticulum/mitochondria contacts. Ann Neurol 78:679–696
Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr, Meyer T (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 15:1235–1241
Loewen CJ, Young BP, Tavassoli S, Levine TP (2007) Inheritance of cortical ER in yeast is required for normal septin organization. J Cell Biol 179:467–483
Lopez Sanjurjo CI, Tovey SC, Taylor CW (2014) Rapid recycling of Ca2+ between IP3-sensitive stores and lysosomes. PLoS One 9:e111275
Lopez-Sanjurjo CI, Tovey SC, Prole DL, Taylor CW (2013) Lysosomes shape ins(1,4,5)P3-evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J Cell Sci 126:289–300
Luik RM, Wu MM, Buchanan J, Lewis RS (2006) The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol 174:815–825
Lur G, Haynes LP, Prior IA, Gerasimenko OV, Feske S, Petersen OH, Burgoyne RD, Tepikin AV (2009) Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP(3) receptors. Curr Biol 19:1648–1653
Maiellaro I, Lefkimmiatis K, Moyer MP, Curci S, Hofer AM (2012) Termination and activation of store-operated cyclic AMP production. J Cell Mol Med 16:2715–2725
Malli R, Frieden M, Osibow K, Zoratti C, Mayer M, Demaurex N, Graier WF (2003) Sustained Ca2+ transfer across mitochondria is essential for mitochondrial Ca2+ buffering, sore-operated Ca2+ entry, and Ca2+ store refilling. J Biol Chem 278:44769–44779
Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M (2012) MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151:630–644
Manford AG, Stefan CJ, Yuan HL, Macgurn JA, Emr SD (2012) ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev Cell 23:1129–1140
Marriott I, Mason MJ (1995) ATP depletion inhibits capacitative Ca2+ entry in rat thymic lymphocytes. Am J Phys 269:C766–C774
Mattiazzi Usaj M, Brloznik M, Kaferle P, Zitnik M, Wolinski H, Leitner F, Kohlwein SD, Zupan B, Petrovic U (2015) Genome-wide localization study of yeast Pex11 identifies peroxisome-mitochondria interactions through the ERMES complex. J Mol Biol 427:2072–2087
McCormack JG, Halestrap AP, Denton RM (1990) Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 70:391–425
Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B (2013) A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 155:830–843
Mesmin B, Bigay J, Polidori J, Jamecna D, Lacas-Gervais S, Antonny B (2017) Sterol transfer, PI4P consumption, and control of membrane lipid order by endogenous OSBP. EMBO J 36:3156–3174
Montalvo GB, Artalejo AR, Gilabert JA (2006) ATP from subplasmalemmal mitochondria controls Ca2+-dependent inactivation of CRAC channels. J Biol Chem 281:35616–35623
Montero M, Alonso MT, Carnicero E, Cuchillo-Ibanez I, Albillos A, Garcia AG, Garcia-Sancho J, Alvarez J (2000) Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat Cell Biol 2:57–61
Morgan AJ, Davis LC, Wagner SK, Lewis AM, Parrington J, Churchill GC, Galione A (2013) Bidirectional Ca(2)(+) signaling occurs between the endoplasmic reticulum and acidic organelles. J Cell Biol 200:789–805
Murley A, Nunnari J (2016) The emerging network of mitochondria-organelle contacts. Mol Cell 61:648–653
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M, Herkenne S, Hernandez-Alvarez MI, Zorzano A, De Stefani D, Dorn GW 2nd, Scorrano L (2016) Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc Natl Acad Sci U S A 113:11249–11254
Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M, Herkenne S, Hernandez-Alvarez MI, Zorzano A, De Stefani D, Dorn GW 2nd, Scorrano L (2017) Reply to Filadi et al.: Does Mitofusin 2 tether or separate endoplasmic reticulum and mitochondria? Proc Natl Acad Sci U S A 114:E2268–E2269
Nunes P, Demaurex N (2014) Redox regulation of store-operated Ca2+ entry. Antioxid Redox Signal 21:915–932
Nunes P, Cornut D, Bochet V, Hasler U, Oh-Hora M, Waldburger JM, Demaurex N (2012) STIM1 juxtaposes ER to phagosomes, generating Ca(2)(+) hotspots that boost phagocytosis. Curr Biol 22:1990–1997
Okeke E, Dingsdale H, Parker T, Voronina S, Tepikin AV (2016) Endoplasmic reticulum-plasma membrane junctions: structure, function and dynamics. J Physiol 594:2837–2847
Orci L, Ravazzola M, Le Coadic M, Shen WW, Demaurex N, Cosson P (2009) From the cover: STIM1-induced precortical and cortical subdomains of the endoplasmic reticulum. Proc Natl Acad Sci U S A 106:19358–19362
Parekh AB (2003) Store-operated Ca2+ entry: dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane. J Physiol 547:333–348
Parekh AB, Putney JW Jr (2005) Store-operated calcium channels. Physiol Rev 85:757–810
Park MK, Ashby MC, Erdemli G, Petersen OH, Tepikin AV (2001) Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J 20:1863–1874
Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D, Rizzuto R (2014) MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53:726–737
Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467:291–296
Phillips MJ, Voeltz GK (2016) Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol 17:69–82
Ping HA, Kraft LM, Chen W, Nilles AE, Lackner LL (2016) Num1 anchors mitochondria to the plasma membrane via two domains with different lipid binding specificities. J Cell Biol 213:513–524
Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK (2013) MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8:e55785
Prinz WA (2014) Bridging the gap: membrane contact sites in signaling, metabolism, and organelle dynamics. J Cell Biol 205:759–769
Qiao X, Jia S, Ye J, Fang X, Zhang C, Cao Y, Xu C, Zhao L, Zhu Y, Wang L, Zheng M (2017) PTPIP51 regulates mouse cardiac ischemia/reperfusion through mediating the mitochondria-SR junction. Sci Rep 7:45379
Quintana A, Pasche M, Junker C, Al-Ansary D, Rieger H, Kummerow C, Nunez L, Villalobos C, Meraner P, Becherer U, Rettig J, Niemeyer BA, Hoth M (2011) Calcium microdomains at the immunological synapse: how ORAI channels, mitochondria and calcium pumps generate local calcium signals for efficient T-cell activation. EMBO J 30:3895–3912
Raychaudhuri S, Prinz WA (2008) Nonvesicular phospholipid transfer between peroxisomes and the endoplasmic reticulum. Proc Natl Acad Sci U S A 105:15785–15790
Rizzuto R, Simpson AW, Brini M, Pozzan T (1992) Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358:325–327
Rizzuto R, Brini M, Murgia M, Pozzan T (1993) Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262:744–747
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280:1763–1766
Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169:435–445
Rowland AA, Chitwood PJ, Phillips MJ, Voeltz GK (2014) ER contact sites define the position and timing of endosome fission. Cell 159:1027–1041
Salo VT, Belevich I, Li S, Karhinen L, Vihinen H, Vigouroux C, Magre J, Thiele C, Holtta-Vuori M, Jokitalo E, Ikonen E (2016) Seipin regulates ER-lipid droplet contacts and cargo delivery. EMBO J 35:2699–2716
Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK (2013) EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342:1379–1382
Schulz TA, Choi MG, Raychaudhuri S, Mears JA, Ghirlando R, Hinshaw JE, Prinz WA (2009) Lipid-regulated sterol transfer between closely apposed membranes by oxysterol-binding protein homologues. J Cell Biol 187:889–903
Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48:158–167
Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu SS (2000) Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J Bioenerg Biomembr 32:97–104
Sheftel AD, Zhang AS, Brown C, Shirihai OS, Ponka P (2007) Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 110:125–132
Shkryl VM, Shirokova N (2006) Transfer and tunneling of Ca2+ from sarcoplasmic reticulum to mitochondria in skeletal muscle. J Biol Chem 281:1547–1554
Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12:2245–2256
Sohn M, Korzeniowski M, Zewe JP, Wills RC, Hammond GRV, Humpolickova J, Vrzal L, Chalupska D, Veverka V, Fairn GD, Boura E, Balla T (2018) PI(4,5)P2 controls plasma membrane PI4P and PS levels via ORP5/8 recruitment to ER-PM contact sites. J Cell Biol 217:1797–1813
Stone SJ, Vance JE (2000) Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem 275:34534–34540
Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R (2006) Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175:901–911
Szalai G, Csordas G, Hantash BM, Thomas AP, Hajnoczky G (2000) Calcium signal transmission between ryanodine receptors and mitochondria. J Biol Chem 275:15305–15313
Tarasov AI, Griffiths EJ, Rutter GA (2012) Regulation of ATP production by mitochondrial Ca(2+). Cell Calcium 52:28–35
Tarasov AI, Semplici F, Li D, Rizzuto R, Ravier MA, Gilon P, Rutter GA (2013) Frequency-dependent mitochondrial Ca(2+) accumulation regulates ATP synthesis in pancreatic beta cells. Pflugers Arch 465:543–554
Tinel H, Cancela JM, Mogami H, Gerasimenko JV, Gerasimenko OV, Tepikin AV, Petersen OH (1999) Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca(2+) signals. EMBO J 18:4999–5008
Valm AM, Cohen S, Legant WR, Melunis J, Hershberg U, Wait E, Cohen AR, Davidson MW, Betzig E, Lippincott-Schwartz J (2017) Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546:162–167
van der Bliek AM, Shen Q, Kawajiri S (2013) Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol 5
Vance JE (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem 265:7248–7256
Vance JE (2014) MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta 1841:595–609
Varadi A, Cirulli V, Rutter GA (2004) Mitochondrial localization as a determinant of capacitative Ca2+ entry in HeLa cells. Cell Calcium 36:499–508
Voronina S, Sukhomlin T, Johnson PR, Erdemli G, Petersen OH, Tepikin A (2002) Correlation of NADH and Ca2+ signals in mouse pancreatic acinar cells. J Physiol 539:41–52
Voronina SG, Barrow SL, Gerasimenko OV, Petersen OH, Tepikin AV (2004) Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. J Biol Chem 279:27327–27338
Voronina SG, Barrow SL, Simpson AW, Gerasimenko OV, da Silva Xavier G, Rutter GA, Petersen OH, Tepikin AV (2010) Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology 138:1976–1987
Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong DW, Stanley WC, Sztalryd C (2011) Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res 52:2159–2168
Wang PT, Garcin PO, Fu M, Masoudi M, St-Pierre P, Pante N, Nabi IR (2015) Distinct mechanisms controlling rough and smooth endoplasmic reticulum contacts with mitochondria. J Cell Sci 128:2759–2765
Willoughby D, Wachten S, Masada N, Cooper DM (2010) Direct demonstration of discrete Ca2+ microdomains associated with different isoforms of adenylyl cyclase. J Cell Sci 123:107–117
Willoughby D, Everett KL, Halls ML, Pacheco J, Skroblin P, Vaca L, Klussmann E, Cooper DM (2012) Direct binding between Orai1 and AC8 mediates dynamic interplay between Ca2+ and cAMP signaling. Sci Signal 5:ra29
Wong YC, Ysselstein D, Krainc D (2018) Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554:382–386
Xu N, Zhang SO, Cole RA, McKinney SA, Guo F, Haas JT, Bobba S, Farese RV Jr, Mak HY (2012) The FATP1-DGAT2 complex facilitates lipid droplet expansion at the ER-lipid droplet interface. J Cell Biol 198:895–911
Zaar K, Volkl A, Fahimi HD (1987) Association of isolated bovine kidney cortex peroxisomes with endoplasmic reticulum. BBA 987:135–142
Zajac AL, Goldman YE, Holzbaur EL, Ostap EM (2013) Local cytoskeletal and organelle interactions impact molecular-motor- driven early endosomal trafficking. Curr Biol 23:1173–1180
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the special issue on Mitochondrial Signalling in Pflügers Archiv-European Journal of Physiology
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tepikin, A.V. Mitochondrial junctions with cellular organelles: Ca2+ signalling perspective. Pflugers Arch - Eur J Physiol 470, 1181–1192 (2018). https://doi.org/10.1007/s00424-018-2179-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-018-2179-z