
Abstract
The hair bundles of cochlear hair cells play a central role in the auditory mechano-electrical transduction (MET) process. The identification of MET components and of associated molecular complexes by biochemical approaches is impeded by the very small number of hair cells within the cochlea. In contrast, human and mouse genetics have proven to be particularly powerful. The study of inherited forms of deafness led to the discovery of several essential proteins of the MET machinery, which are currently used as entry points to decipher the associated molecular networks. Notably, MET relies not only on the MET machinery but also on several elements ensuring the proper sound-induced oscillation of the hair bundle or the ionic environment necessary to drive the MET current. Here, we review the most significant advances in the molecular bases of the MET process that emerged from the genetics of hearing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The ability of vertebrates to maintain their balance and sense sound vibrations is decisive for their survival. Although vertebrates live in various environments, they all make use of the same organelle, the hair bundle, that transduces mechanical information into an electrical signal in sensory hair cells. Hair cells are present in the neuromasts of lateral lines in fish and amphibian larvae, where they detect water movement; in the vestibular end organs, where they detect linear and angular acceleration; and in the auditory organs, where they detect sound pressure waves (Fig. 1a). Hair cells are also present in non-vertebrate organisms. For instance, the sea anemone, which belongs to the cnidarian phylum, uses hair cells located on its tentacles to detect zoo-plankton [228, 229]. The hair bundle is located at the apex of hair cells and is comprised of several rows of rigid, actin-filled microvilli, known as stereocilia, which are organised in a staircase pattern and maintained together by different types of links. One link, called the tip link, plays a major role in mechano-electrical transduction (MET). This oblique link connects the tip of each sterocilium to the lateral wall of the adjacent taller stereocilium. Upon mechanical stimulation of the hair bundle in the direction of the tallest stereocilia, i.e. the excitatory direction, tension in the tip links increases resulting in a higher probability of MET channel opening and cell depolarisation [87, 86, 164, 67, 10] (Fig. 1b, c). The biophysical features of these MET channels have been extensively studied. These cationic non-selective channels [23, 41, 154] have a large unitary conductance in the 100 pS range [43, 71, 154] and an extremely fast activation time constant [42, 205, 172], and are permeant to large organic cations such as choline and TEA [62, 154].

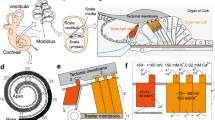
Auditory organ and MET. a Schematic cross-section of the cochlea. IHC inner hair cell, OHC outer hair cell. b Illustration of the stimulation of a mature OHC hair bundle. Stereocilia are maintained cohesive by top connectors (purple). The tallest row of stereocilia is anchored into the tectorial membrane. Upon hair bundle displacement towards this row (excitatory direction), high tension in the tip-links results in MET channel opening, leading to the entry of K+ and Ca2+ ions to the hair cell. c Left Example of transduction current recordings in an IHC, voltage clamped at −80 mV, of a P7 mouse while applying different displacement steps with a glass probe in the excitatory direction and a 180-nm step in the inhibitory direction (calibrated voltage command of the stimulator at the top left). Right Corresponding current–displacement plot fitted with a three-state Boltzmann relation
The first physical description of hair bundle functioning was reported in the late 1970s. However, the small number of hair cells in the inner ear (a few thousands) hampered molecular advances, as opposed to other sensory organs like the eye, which contains more than 100 million photoreceptor cells. In the early 1990s, human genetics, the efficiency of which is independent from the number of hair cells, emerged as the best approach to identify molecules involved in MET. Studies focused largely on the cochlea, the mammalian auditory organ, rather than on the vestibular organs because vestibular defects in humans are often compensated by the visual and proprioceptive systems. In addition, deafness is the most frequent sensory defect at birth (approximately one out of 700 newborns is affected by severe or profound hearing impairment). Currently, more than 120 deafness loci have been characterised, and around 80 genes responsible for isolated (non-syndromic) forms of sensorineural deafness have been identified (see the Hereditary Hearing Loss Homepage: http://hereditaryhearingloss.org). In addition, many more genes are involved in syndromic forms of sensorineural deafness. Pathophysiological studies rely on multidisciplinary approaches that include invasive exploration techniques in animal models. Mouse models offer substantial possibilities for genetic manipulation and have proven to be highly relevant for the understanding of human auditory defects because mutations in mouse orthologues of the genes associated with deafness in humans faithfully mimic the sensory defect in most cases.
The auditory sensory epithelium of mammals, which is called the organ of Corti (Fig. 1a), is comprised of the hair cells and of various types of supporting cells that are sandwiched between the underlying basilar membrane and the overlying tectorial membrane. Upon sound stimulation, the shearing movement between the basilar membrane and the tectorial membrane deflects the hair bundles of hair cells at the frequency of the stimulus. Each hair cell along the cochlear longitudinal axis is tuned to be highly sensitive to a particular frequency, called its characteristic frequency. Together, the hair cells form a tonotopic map from the base to the apex of the cochlea. There are two types of hair cells in the cochlea: the inner hair cells (IHCs), which are organised in one row, and the outer hair cells (OHCs), which are organised in three rows (Figs. 1a and 2a). IHCs are the genuine sensory cells that transduce the sound stimuli into an electrical signal in the primary auditory neurons, whereas OHCs carry out frequency dependent mechanical amplification of sound-evoked vibrations of the organ of Corti.

Hair bundle polarity and morphology. a Left Scanning electron micrograph (SEM) of the organ of Corti in a P6 wild-type mouse. The U- and V-shaped hair bundles of IHCs and OHCs are aligned and their vertices point towards the cochlear abneural edge. Scale bar, 5 μm. Right Examples of OHC and IHC hair bundles in a P6 wild-type mouse. Scale bars, 1 μm. b Left SEM of OHC hair bundles in a sans-null mutant mouse at P5 (Jackson shaker). Right SEM of a basal IHC hair bundle in a sans cKO (Myo15-cre +/−x Ush1g fl/fl) mouse and in a control mouse at P8. The white frame highlights the presence and absence of prolate shapes of representative stereocilia tips for the control and the cKO genotype, respectively. Scale bars, 1 μm. c Left SEM of IHC hair bundles in a myo7a-null mosaic mouse mutant. In this mouse, Myo7a was expressed transgenically on the X-chromosome of myo7a-null mutants, enabling direct comparison, within the same organ of Corti, between myosin VIIa-deficient (single asterisk) and -complemented (double asterisk) hair cells due to random X-chromosome inactivation among hair cells [167]. Note that the stereocilia of the tallest row are longer in the myosin VIIa-deficient (single asterisk) IHC than in the myosin VIIa-complemented (double asterisk) IHC. Right SEM of an IHC hair bundle in a whirlin-null (whirler) mouse and in a control mouse. Note the abnormally short stereocilia in the whirlin-null IHC; as a result, the kinocilium (arrowhead) is taller than the stereocilia. Scale bars, 1 μm
As more and more genes involved in MET are identified, a major challenge is to elucidate the physiological roles of the encoded proteins. More than 80 molecules have already been shown to be essential to MET (see Table 1). However, only a small proportion of these molecules have been identified as components of the MET machinery, based on electrophysiological data and relevant biophysical models. In particular, the molecular identity of the MET channel is still a matter of debate. The molecular motor myosin-VIIa was the first ‘deafness’ gene to be discovered [231, 72]; however, its role in auditory transduction and in particular, its role as a molecular conveyor and as a mechanical tensor has not yet been clarified. Some molecules play several roles at different positions in the hair bundle or at different stages in the development of the transduction apparatus [117, 35]. For instance, abnormal morphogenesis of the hair bundle in knock-out mice defective for such proteins may mask subsequent morphological or functional defects arising at late stages of development. Delayed conditional knock-outs in specific cochlear cell types are useful to examine the possible role of these molecules in the mature hair bundle [35, 160].
Any defect of the hair bundle is expected to have an effect on MET, including defects of hair bundle development, the tectorial membrane, which is involved in its deflection, the endocochlear fluid homeostasis, or the MET machinery itself. In this review, we examine knowledge gathered through neurogenetics regarding the molecules involved in these four aspects of hair bundle functioning, and discuss alternative strategies to complete the molecular picture of molecules involved in MET.
Hair bundle development
Positioning and orienting the hair bundle
Unlike humans that can detect sounds from the sixth month of embryonic development, mice start to hear on postnatal day 12 (P12) because their cochlear sensory epithelium continues to develop after birth. At birth, the first steps of hair bundle growth have already occurred. All the V- or U-shaped hair bundles are aligned, and their vertices point towards the cochlear abneural edge (see [131, 61] for review) (Fig. 2a). Planar polarisation of the hair bundles is essential for their coordinated deflection upon sound stimulation. Between embryonic day 14.5 (E14.5) and E15.5, a specialised primary cilium called the kinocilium, emerges at the centre of the hair cell apical surface, surrounded by microvilli, and migrates towards the cell’s abneural edge. Microvilli then grow differentially in a staircase pattern, eventually forming three stereocilia rows of increasing height. The position of the kinocilium marks the vertex of the hair bundle. Therefore, mutations in genes involved either in planar cell polarisation (PCP) or in kinocilium migration are expected to affect the final polarity of the hair bundle (see Table 1).
Core PCP molecules were originally identified from studies on Drosophila. Vangl2 was the first orthologous gene to be implicated in the orientation of the hair bundle in the mouse [143]. Vangl2 Lp/Lp mutants have normally shaped, but misoriented hair bundles. Defects in several other core PCP molecules including vangl1 [211], frizzled-3 [226], frizzled-6 [226], and disheveled-1, disheveled-2 and disheveled-3 [225, 58] also result in abnormally oriented hair bundles. These core PCP molecules are asymmetrically distributed within the cell and are mostly located at the junctions between hair cells and supporting cells. For example, vangl2 is highly abundant at the adherens junction on the supporting cell side [73, 227]. Mutations in non-core PCP genes including Cthrc1 [239], Ror2 [239], Scrib [143], PTK7 [123, 158, 115], Fat4 [180], Dchs1 [128], Sec24b [139], Smurf1 and Smurf2 [148] also result in hair bundle misorientation. Mutations in these genes give rise to variable phenotypes that are usually less severe than those of mutations in the core PCP genes. Mutations in the genes causing ciliopathies, which are syndromes that result from defects of the primary cilium, also lead to defects of hair bundle polarity. They include some of the genes responsible for Bardet–Biedl syndrome (BBS1 [175], BBS4 [175], MKKS (BBS6) [175] and TTC8 (BBS8) [132]) (see [66] for review), genes responsible for Meckel–Gruber syndrome (Mks1 [44]), and genes responsible for Alström syndrome (Alms1 [89]). The conditional knock-out of genes involved in intraflagellar transport, Ift88 or Kif3a, results in loss of the kinocilium and is associated with PCP defects in mice, providing further evidence for the involvement of the kinocilium in hair bundle orientation [95, 200].
GTP-binding protein αi subunits (Gαi) control mitotic spindle orientation and are associated with GPSM2, which is a protein implicated in deafness [221, 53]. Gαi subunits were recently found to be involved in kinocilium migration and in hair bundle shape and orientation [60, 206]. These proteins are located in the apical region of the hair cell on its abneural side, between the cell junction and the hair bundle, forming a crescent-shaped domain. The role of Gαi in hair bundle shape was confirmed in Gαi3 mutant mice that display flattened hair bundle shapes and mislocalised kinocilia [60]. A complementary domain to that of Gαi at the apical surface of hair cells on the neural side of hair bundles is also defined by the expression of atypical protein kinase C (aPKC) [60, 206]. Thus, the boundary between the Gαi- and aPKC-containing areas may participate in defining the apical surface subregion where the hair bundle emerges [60, 206].
The hair bundle, a cohesive structure
The formation of the hair bundle and the maintenance of its cohesiveness are orchestrated by several types of links that come into play at different developmental stages. Prior to their molecular description, these links were categorised according to both their location and sensitivity to proteases/calcium chelators (Fig. 3) [19, 75]. In the newborn mouse (P0), numerous interstereociliary lateral links interconnect stereocilia across and between rows in different directions. From P2 onwards, three types of lateral links take over, namely ankle links that are located at the base of stereocilia and shaft connectors that are located along stereocilia, and kinocilial links that connect the kinocilium to adjacent stereocilia of the tallest row. In mature cochlear hair cells, only the tip links remain, together with putative lateral links in IHCs and apical top connectors in OHCs [75]. Several molecular components of these links have been identified (see Table 1). Mutations in the corresponding genes in mice lead to congenital hearing impairment and hair-bundle disorganisation, indicating that each link type contributes critically to the building or the maintenance of the hair bundle.

Hair bundle cohesion. Top Schematic illustration of the different types of links between stereocilia in OHCs at three different developmental stages, E17.5, P5, and P14. Bottom Molecular composition of the different links and their associated molecular complexes. Single asterisk The positions of the listed proteins at the upper or lower tip-link insertion points are detailed in Fig. 5. Double asterisk Usherin and PTPRQ are part of the ankle link complex and the shaft link complex, respectively, but it is unknown whether these proteins form the links
The study of the genes responsible for Usher syndrome has been especially informative for our understanding of hair-bundle development. Usher syndrome (USH) is an autosomal recessive disorder that associates congenital hearing impairment with delayed onset retinitis pigmentosa eventually leading to blindness. This disorder has three clinical subtypes. USH1, the most severe form, is characterised by severe to profound congenital deafness, constant vestibular dysfunction and prepubertal onset retinitis pigmentosa. By contrast, USH2 involves only moderate to severe hearing impairment and no vestibular dysfunction. USH3 is distinguished from USH2 by the progressiveness of the hearing impairment and the occasional presence of vestibular dysfunction (see [163] for review). Six genes have been implicated in USH1, three in USH2 and one in USH3. USH1 has been associated with mutations in the genes encoding cadherin-23 (USH1D) [29, 31], protocadherin-15 (USH1F) [6, 8], the PDZ domain-containing protein harmonin (USH1C) [216, 25], the ankyrin repeat- and sterile α motif-containing protein sans (USH1G) [232] (Fig. 2b), the unconventional myosin myosin-VIIa (USH1B) [231] and the calcium and integrin-binding protein CIB2 (USH1J) [171]. USH2 has been associated with mutations in two genes encoding proteins containing a long extracellular domain, the very large G-coupled receptor (VLGR1) (USH2C) [233] and the transmembrane protein usherin (USH2A) [59], and with mutations in the gene encoding the PDZ domain-containing protein whirlin (USH2D) [57]. The gene encoding the four-transmembrane domain protein clarin-1 (USH3A) is the only identified gene associated with USH3 [91, 3, 64]. Genetics brought the first evidence that proteins involved in the various genetic forms of each Usher clinical subtype interact in vivo [26, 117]. In vitro binding experiments then demonstrated their direct interaction. These proteins are either components of the interstereociliary links or are submembrane scaffold proteins that presumably participate in the anchoring of these links to the actin cytoskeleton (Fig. 3). For instance, early transient lateral links, kinocilial links and tip links are made of cadherin-23 and protocadherin-15 [26, 75, 142, 198, 201, 5, 99]. Cadherin-23 forms a ternary complex with harmonin and myosin-VIIa [16]. Protocadherin-15 binds to myosin-VIIa [194] and binds to harmonin in vitro [2, 170]. Mutations in any of the mouse USH1 orthologous genes lead to cochlear hair bundle fragmentation, highlighting their role in hair bundle cohesion as early as E17 [109, 72, 234, 51, 7, 92, 104]. Moreover, the hair bundles of these mutant mice have mispositioned kinocilia and are misoriented [117]. Ankle links are composed of VLGR1 and possibly usherin [1, 136, 140]. These proteins interact with whirlin [214, 1] and PDZD7 [77, 250] that is encoded by a modifier gene of the USH2 phenotype [56]. In Vlgr1 knock-out mice, ankle links are absent and abnormally shaped hair bundles are apparent at P2 [136, 238] (Fig. 3). Paradoxical MET currents can be elicited in these bundles if the stereocilia are deflected in the inhibitory direction by a glass pipette, indicating a lack of hair bundle cohesiveness [140]. In addition, two proteins that are implicated in isolated deafness but not in USH also play a role in hair bundle cohesiveness: tyrosine phosphatase receptor Q (PTPRQ) that is associated with shaft connectors [74, 149] and stereocilin that is associated with OHC top connectors [217, 218].
Control of stereocilia length
Stereocilia are filled with a large core of parallel, densely packed, cross-linked actin filaments with barbed ends at their tips, where actin monomers are incorporated, and with pointed ends at their base, where depolymerisation occurs. Stereocilia taper at their base, which contains fewer actin filaments than the core. These filaments are densely packed to form an array that extends below the apical cell surface, forming the stereocilia rootlets. These rootlets anchor the stereocilia in the cuticular plate, which is a dense meshwork of actin filaments lying beneath the apical surface of the hair cell. The biophysical properties of MET strongly rely on the correct formation and maintenance of the hair bundle staircase pattern.
The shape of stereocilia reflects that of its cytoskeleton, which in turn depends on different categories of actin-interacting proteins. These include (1) actin-nucleating proteins that promote initiation of actin polymerisation, (2) actin-capping proteins that prevent the barbed end from incorporating actin monomers, (3) actin-bundling proteins that cross-link parallel actin filaments, (4) actin side-binding proteins, (5) actin-monomer-sequestering proteins, (6) actin-severing proteins that split actin filaments and (7) actin molecular motors. Mutations in various actin and actin-interacting proteins of these categories cause defects in stereocilia structure (see Table 1). Stereocilia contain β-actin (actb) and γ-actin (actg1), and mutations in ACTG1 and ACTB lead to deafness [81, 144, 161, 166, 213, 249]. Mutations in Diaphanous-1, which encodes an actin-nucleating protein that controls actin polymerisation, cause deafness [125]. Overexpression of Diaphanous-3 also results in deafness due to larger than normal stereocilia [189]. Espin, an actin-bundling protein, is necessary for the assembly and stabilisation of parallel actin filaments. Stereocilia morphogenesis is markedly impaired in the Jerker mutant mouse, which lacks functional espin [248, 150]: as early as P0, stereocilia are abnormally thin and short, with impaired differential elongation that causes the loss of the staircase pattern [191]. Mutations in EPS8, which encodes an actin-bundling and actin-capping protein, cause profound congenital deafness [20]. Eps8 is located predominantly at the tips of stereocilia. In knock-out mice lacking eps8, stereocilia are abnormally short but are still organised in a staircase pattern [244]. Notably, a related actin-bundling and actin-capping protein, eps8-l2, is required for the maintenance of the hair bundle staircase pattern [68]. Radixin (rdx), which belongs to the family of ezrin/radixin/moesin (ERM) proteins, tethers actin filaments to the plasma membrane at the base of stereocilia. Accordingly, mutations in RDX are responsible for hearing impairment in humans [101], and loss of Rdx in mice causes progressive degeneration of stereocilia [107]. NHERF1 and NHERF2, which both contain an ERM binding domain and two PDZ domains [54], have also been implicated in deafness in mice [96]. NHERF2 is mainly located at the base of hair bundles of cochlear hair cells and is more abundant in IHCs than in OHCs [196, 96]. NHERF1 is present in the hair bundles of both IHCs and OHCs at embryonic stages before concentrating at the stereocilia tips of OHCs and could possibly bind to cadherin-23 in vivo [96]. In Nherf1 −/− mice, the hair bundles of OHCs have abnormal shapes in the basal and middle cochlear regions. Interestingly, this tonotopy-dependent phenotype has revealed an unusually powerful mode of interference between low- and high-frequency sounds, suggesting a previously unreported mode of off-frequency hearing [96]. Studies involving Triobp mutant mice, which lack both TRIOBP-4 and TRIOBP-5, show that the actin-bundling protein TRIOBP is necessary for the formation of stereocilia rootlets [108]. Many other actin-interacting proteins have been detected in stereocilia including the actin side-binding protein tropomyosin [69], the actin-severing protein cofilin [146] and the actin-bundling proteins fimbrin [210] and fascin-2 [162].
The hair bundle also contains various unconventional myosins. Their respective contributions in molecular transport and in the maintenance of mechanical tension have not yet been clarified. Myosins are logical candidates to transport proteins along the stereocilia dense network of actin filaments [209]. Moreover, their presence at different locations, especially near the tip or at the base of stereocilia, may exert tension on actin filaments and modify stereocilia shape. The study of myosin-IIIa, myosin-VI, myosin-VIIa and myosin-XV has provided additional information about the molecular complexes involved in the maintenance of the stereocilia actin cores (Fig. 4). Myosin-IIIa [222] accumulates at stereocilia tips [188, 223] and promotes stereocilia lengthening when overexpressed with espin-1 in hair cells [184]. Stereocilia grow excessively and fuse together in mutant mice deficient for myosin-VI [14, 13, 193]. It has recently been proposed that myosin-VI participates in a molecular complex with CLIC5, PTPRQ, radixin and taperin, which are all present at the base of stereocilia [183, 70]. This complex may help to stabilise interactions between the plasma membrane and the subcortical actin cytoskeleton, which may explain the fusion of stereocilia in myosin-VI-deficient mice [182, 183]. Nonetheless, the mechanism of stereocilia overgrowth in these mice is still poorly understood. The tallest row of stereocilia in mutant mice deficient for myosin-VIIa is also abnormally long [167] (Fig. 2c). This phenotype has been ascribed to the concomitant loss of twinfilin-2, an actin-sequestering and actin-capping protein that inhibits actin polymerisation [178, 159]. Another molecular complex was uncovered by the observation of abnormally short stereocilia in myosin-XV-defective [165] and whirlin-defective mouse mutants [134] (Fig. 2c). Myosin-XV and whirlin interact and form a complex with eps8 that plays a crucial role in the elongation of the stereocilia actin filaments [50, 21, 127]. This complex also includes the membrane-associated guanylate kinase (MAGUK) p55, protein 4.1R [133] and gelsolin, which is an actin-capping and actin-severing protein [135]. Therefore, several myosin-dependent molecular complexes that are linked to actin dynamics work in concert to determine stereocilia length.

List of myosins and their interactors involved in the control of stereocilia length. The roles of myosin-VI, myosin-VIIa, and myosin-XV have been determined by the study of mutant mice defective for these proteins. In contrast, the implication of myosin-IIIa in stereocilia elongation was assessed in vitro from the observation that stereocilia are taller than normal in co-transfected hair cells producing myosin-IIIa and espin 1 [184]. Single asterisk These proteins have not been associated with deafness forms in humans or in mice
The molecular processes that determine stereocilia differential elongation in different rows are still unknown. However, several studies, with conflicting results, have addressed the issue of steady-state actin renewal in mature hair bundles. A treadmilling process was first proposed to ensure the renewal of actin monomers in stereocilia filaments. When actin fused to the green fluorescent protein (actin-GFP) was overexpressed in cells, the actin core renewal speed was unexpectedly fast (~48 h) [187], and turnover time was similar in different stereocilia rows. This implies an approximate proportional relationship between stereocilia size and the speed of actin polymerisation [179]. However, the overexpression of a modified actin monomer (actin-GFP) might alter the intrinsic properties of actin in stereocilia. An alternative approach based on the incorporation of 15N-labelled precursor amino acids by multi-isotope imaging mass spectrometry in stereocilia indeed suggested otherwise, i.e. that the overall protein renewal including actin is slow (around 10 days in young mice and 50 days in adult mice) and faster at the very tip (distal 0.5 μm end) than in the core of stereocilia [245]. However, the time resolution in this radio-labelling approach is limited by the life time of proteins, which might be much longer than the local turnover time of actin filaments by a treadmilling process.
The mature MET apparatus
The MET machinery
High-speed imaging of the calcium influx through MET channels in cochlear hair cells has shown that these channels are located at the tips of the short and middle row stereocilia but not in tall row stereocilia. MET channels would therefore be located at the basal ends of the tip links [24]. The molecular nature of the MET channel has so far remained elusive. The transmembrane channel-like 1 (TMC1) and TMC2 proteins, which have six transmembrane domains, are currently the best candidates. Indeed, mutations in TMC1 cause deafness in humans [113] and inner ear hair cells from double knock-out mice for Tmc1 and Tmc2 have no MET currents [98]. In addition, the re-expression of various combinations of Tmc1, Tmc2, and mutated forms of Tmc1 in the hair cells of these double knock-out mice [157] modifies the single MET channel conductance and its permeability to Ca2+ ions. This suggests that TMC1 and TMC2 are pore-forming subunits of the MET channel [157, 106]. However, this view was recently challenged by the observation that a mechano-sensitive current could still be elicited in the double knock-out mice by pushing the hair bundle in the inhibitory direction [105]. Therefore, TMC1 and TMC2 may not constitute the MET channel by themselves, but instead may be essential for its targeting to the stereocilia tips (see [18] for comment and see [83] for review). A recent study revived the debate by showing that the ion channels underlying the anomalous MET current elicited by pushing the hair bundle in the inhibitory direction may in fact have pore properties different from those of the genuine MET channels, based on the lower dihydrostreptomycin-blocking efficacy and the absence of rectification in their current–voltage relationship [129].
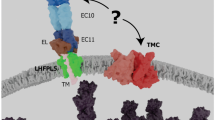
The upper and lower parts of the tip link are composed of cadherin-23 (USH1D) and protocadherin-15 (USH1F), respectively [198, 201, 5, 99]. Inner ear hair cells express three different transmembrane protocadherin-15 isoforms, CD1, CD2 and CD3, that differ in their intracellular amino acid sequence [5]. Based on the study of knock-out mice, each of them being defective for only one protocadherin-15 isoform, it has been suggested that protocadherin15 isoforms are functionally redundant [230]. However, the analysis of a delayed, hair cell-specific conditional knockout mouse that loses only the protocadherin-15-CD2 isoform after the period of hair-bundle development has shown that this isoform is an essential component of the tip link in mature auditory hair cells [160]. In addition, a PCDH15 mutation that affects only the CD2 isoform was also found to lead to profound deafness without vestibular defects in human patients. Because mutant mice for CD1 or CD3 are not hearing-impaired [230], CD2 would be the only isoform of protocadherin-15 required for the tip link in mature IHCs and OHCs, unless CD1 and CD3 are functionally redundant [160]. Three other USH1 proteins, harmonin, sans and myosin-VIIa, and a non-USH gene, tetraspan membrane protein of hair cell stereocilia (TMHS), have been shown to participate in molecular complexes associated with the lower and upper tip-link insertion points (Fig. 5). Harmonin isoforms comprise three sub-classes: a, b, and c. The largest isoform, harmonin-b, that contains three PDZ domains, two coiled-coil domains and one PST domain, is an F-actin-bundling protein [26] and is located at the upper tip-link insertion point in the mature hair bundle [117, 141, 79]. Electrophysiological studies of MET currents in cochlear explants of harmonin-b null mice are consistent with a role of this protein as an internal linker between the tip link and the actin cytoskeleton [141]. The contributions of isoforms a and c to MET are still unclear [26, 79, 141]. Sans, which binds to harmonin [2, 240] and myosin-VIIa in vitro [2, 235], and possibly to the intracellular domains of cadherin-23 and protocadherin-15, is located at the lower tip-link insertion point in the developing hair bundle [35] and at the upper tip-link insertion point in the mature hair bundle [76]. Late conditional knock-out (after the development of the hair bundle) of the sans gene in hair cells results in markedly impaired transduction currents [35]. This has been ascribed to the loss of the tip links, implying that sans is necessary to maintain the tip link in the mature MET machinery. The involvement of myosin-VIIa in MET is likely to be more complex than that of sans since this motor protein probably has several functions. Mutant mice defective for myosin-VIIa have severely damaged hair bundles [192]. This myosin interacts with most of the other USH proteins and may intervene in their transport in the hair bundle, which may explain this phenotype. For instance, in myosin-VIIa-defective mouse mutants, two major components of the ankle-link complex, VLGR1 and usherin, are absent from the hair bundle as well as harmonin-b, but not cadherin-23 [140, 117, 194]. In the mature hair bundle, myosin-VIIa is observed in the region of the upper tip-link insertion point [76], where it is expected to form a ternary complex with harmonin-b and cadherin-23 as it does in vitro [16]. All USH1 proteins identified so far are involved in the MET machinery (Fig. 5), although the role of CIB2 has not yet been defined [171]. Finally, TMHS, a non-USH gene responsible for an autosomal recessive form of deafness, encodes a four-transmembrane domain protein that is located at the lower tip-link insertion point. TMHS binds to protocadherin-15 in vitro. Tmhs knock-out mice have very weak MET currents. However, this phenotype is partially rescued by the overexpression of protocadherin-15, indicating that impaired MET was mostly due to the defective recruitment of this protein. This suggests that TMHS is a key component of the MET machinery, possibly bridging protocadherin-15 to the MET channel, but is not a component of the MET channel itself [236] (Fig. 5).

The MET machinery in cochlear hair cells. a In the developing hair bundle, the MET machinery comprises the MET channel(s) and TMHS at the lower tip-link insertion point. Sans and myosin-VIIa are also present, but the nature of their interaction with the MET complex is still unknown. The nature of the interaction between the MET complex and the actin cytoskeleton is also unknown at the lower tip link insertion point. At the upper tip-link insertion point, myosin-VIIa and harmonin b interact with cadherin-23. The role of myosin-Ic remains unclear in the cochlea because its function has not yet been tested in mice mutant for this protein. In addition, the location of myosin-Ic cannot be investigated by immunohistochemistry due to the absence of the appropriate mutant mice to confirm the specificity of antibodies directed against this protein. b Mature MET machinery. Sans, myosin-VIIa and harmonin-b are located at the upper tip-link insertion point
The MET machinery, a structure under tension
Several features indicate that the MET machinery is subjected to tension even in the absence of sound stimuli. Stereocilia tips of short and middle rows have a prolate shape that is thought to be caused by the resting tension exerted by the tip link on the plasma membrane (Fig. 2b). Direct recordings of receptor potentials in cochlear hair cells in response to sound stimulation in vivo, and of MET currents in response to displacement of the hair bundle in vitro, have shown that a proportion of MET channels are open at rest [42, 45, 176, 94]. This suggests that the resting tension applied to the MET machinery is tightly controlled. This tension is perturbed in several mouse mutants involving molecules of the MET machinery. The phenotypic consequences of conditional knock-out of the sans gene appear at P8 and involve the simultaneous loss of tip links and of the prolate shape of IHC stereocilia tips (Fig. 2b). The prolate shape of stereocilia tips is also absent in cadherin-23 conditional knock-out mice that display an abnormal phenotype involving mature hair cells (beyond P23). Interestingly, in these two models, the loss of the prolate shape is concomitant with the regression of stereocilia in the short and middle rows [35, 34]. These observations are consistent with the hypothesis that tip-link tension controls actin polymerisation at the barbed end of stereocilia actin filaments [168].
The control of the holding tension on the MET machinery depends on the anchoring of the MET channel and the tip link to the actin cytoskeleton. The tip-link tension can be modulated by sliding of the tip-link upper end anchoring point along actin filaments. This mechanism is thought to contribute to the adaptation process that is reflected in the decline in the transduction current evoked by a step displacement of the hair bundle in vitro [85, 9, 55, 111]. Myosins, which are actin-based motors, are natural candidates for the control of tip-link tension by this mechanism. A chemical–genetic strategy in the mouse indeed provided support for a critical role of myosin-Ic in the MET adaptation process in vestibular hair cells [82, 204]. However, it remains unclear which myosin(s) are involved in cochlear hair cells. Myosin-VIIa, which is present at the tip-link upper insertion point in mature cochlear hair cells, is a promising candidate for the MET machinery. However, the role(s) of myosin-VIIa in MET remain(s) unclear because hair bundles in the mutant mice defective for myosin-VIIa are strongly disorganised, making it difficult to attribute the abnormal functional features to a malfunctioning of the MET machinery only. Moreover, MET currents observed in Myo7a 4626SB mice show characteristics similar to the abnormal currents observed in TMC1 and TMC2 defective mutants when hair bundles are pushed in the inhibitory direction, which suggests that the recorded MET currents in Myo7a 4626SB mice would not be gated by tip links (see above) [110, 129]. The b isoform of harmonin also participates in the anchoring of the tip-link upper end to the actin cytoskeleton. In mutant mice that only lack this isoform, MET currents display a variable extent of adaptation. This observation is consistent with a role of harmonin-b as a component of the “extent spring” [195], a mechanical element that has been postulated to control the stroke of the myosin motors in the adaptation process [141]. The dynamic interplay between myosin-VIIa and harmonin-b, both of which can bind to actin at the upper tip-link end, still has to be elucidated. At the lower tip-link insertion point, little is known about the molecules that anchor the MET machinery to the actin cytoskeleton, even though several myosins are present at the stereocilia tips, including myosin-IIIa, myosin-IIIb [138] and myosin-XV (see above).
The tectorial membrane
In the cochlea, hair bundles are covered by an acellular gel composed of several types of collagen and non-collagenous glycoproteins called the tectorial membrane. Like the organ of Corti, the tectorial membrane runs along the cochlear duct. It is attached on its medial side to the spiral limbus, and on the other side, it is in firm contact with the tips of the tallest OHC stereocilia row. Notably, hair bundles of IHCs are free standing under the tectorial membrane. Upon sound stimulation, the shear movement between the basilar membrane and the tectorial membrane drives hair bundle oscillations. Many proteins involved in the composition of the tectorial membrane or required for its attachment to hair cells are encoded by genes associated with deafness. The study of mice mutant for these genes has shed new light on the different roles played by the tectorial membrane in auditory MET.
Six non-collagenous glycoproteins have been found in the tectorial membrane: α-tectorin, β-tectorin, otogelin, otogelin-like, CEACAM16, and otolin [173, 121, 37, 199, 208, 247, 243, 30, 46, 97] (see Table 1 for deafness genes). Notably, the targeted mutation of α- and β-tectorin in mice has helped to characterise the mechanical properties of the tectorial membrane. The bulk of the tectorial membrane is made of several collagen fibres that are organised into a matrix composed of α- and β-tectorins. Inactivation of the α-tectorin gene in Tecta ΔENT/ΔENT mice, which causes the tectorial membrane to detach from the surface of the organ of Corti, led to the conclusion that the elasticity of the tectorial membrane has little influence on the amplitude and phase of deflexion of OHC stereocilia at the characteristic frequency. Rather, at this frequency, the tectorial membrane probably behaves mostly as an inert mass on which OHC stereocilia can react, ensuring that the OHCs respond to sound stimulation with the proper gain and timing [119]. Subsequently, the study of a knock-in mouse harbouring the semi-dominant Tecta Y1870C mutation pinpointed a second mechanical role of the tectorial membrane. Although OHC MET activity is normal in Tecta Y1870C/+mice, neural thresholds are markedly high, indicating that the tectorial membrane also plays a critical role in driving the hair bundles of IHCs [120]. Three knock-in mouse lines with different missense mutations that change amino acid residues in distinct protein subdomains of α-tectorin have recently been produced. The analysis of these mice showed that these subdomains, when defective, affect the biomechanical properties of the tectorial membrane in different ways [118]. A third mechanical role has also been attributed to the tectorial membrane. The striated sheet formed by the two tectorins is disrupted in knock-out mice for the β-tectorin gene (Tectb −/− mice). Basilar membrane and neural tunings are both sharper than normal in these mice, suggesting that the tectorial membrane also influences the longitudinal spread of sound-induced excitation along the cochlea [177]. Several molecules involved in the two main attachments of the tectorial membrane have also been characterised. Otoancorin, which is present at the apical surface of the spiral limbus, plays a critical role in the attachment of the tectorial membrane to this structure. In otoancorin knock-out mice, the tectorial membrane is still attached to the OHC stereocilia but detaches from the spiral limbus, leading to the defective stimulation of IHCs [124]. Notably, the OHC response in these mutants is largely unaffected, despite the concomitant detachment of the TM from the spiral limbus. This reinforces the hypothesis that the elasticity of the tectorial membrane plays little role in the stimulation of OHCs near their characteristic frequency. Stereocilin is an extracellular protein of the mature OHC hair bundle. Top connectors do not form in stereocilin knock-out mice, and stereocilia imprints do not appear on the tectorial membrane. Thus, stereocilin is necessary for the formation of top connectors, and it may be a component of the “attachment links” that connect the tallest stereocilia of OHCs to the tectorial membrane. Whether these attachment structures are formed by genuine fibrous links or by the extracellular matrix remains unclear. The absence of the top connectors leads to deafness caused by progressive disorganisation of the hair bundle, which is preceded by a loss of the acoustic distortion products normally generated by OHC hair bundles [218, 215] (see [11] for review).
Ionic composition of the endolymph
Hair bundles are bathed in endolymph, which is an extracellular fluid with an unusually high K+ concentration (approximately 150 mM [185]). There is a +80–100 mV transepithelial potential difference between the endolymphatic and perilymphatic compartments (endocochlear potential) [137, 153, 186]. The resulting 120–150 mV difference between the endolymph and the intracellular compartment [94] drives the MET current, mainly carried by K+ ions, into the hair cells. The endocochlear potential and the high K+ concentration of the endolymph are produced by the stria vascularis, a specialised bi-layered epithelium of the cochlear duct outer wall. The maintenance of the endocochlear potential requires the integrity of the cell–cell tight junctions that keep the endolymphatic and perilymphatic compartments electrically isolated from one another. Several ion channels and transporters have been implicated in the production of the endocochlear potential and/or K+ secretion by the stria vascularis, including the Kcnj10 [130, 242], Kcnq1 [151, 116], and Kcne1 [219, 212, 190] K+ channel subunits, and the Na+–K+–2Cl− cotransporter NKCC1 [49, 52]. Loss-of-function mutations in any of these genes result in severe hearing impairment.
The existence of a recycling, through an intercellular gap junction network, K+ ions that flow out of the hair cells in their basolateral region has been suggested, although such a process remains to be established. Mutations in the connexin 26 gene (CX26/GJB2) [100] are the most common cause of autosomal recessive congenital deafness in many Caucasian populations; however, the various roles of gap junction channels in the functioning of the cochlea are still poorly understood. The conditional knock-out of Gjb2 in the mouse organ of Corti leads to the degeneration of sensory cells and supporting cells. This phenotype has been attributed to defects in the gap junctions that would be involved in the recycling of K+ ions released at the base of hair cells. In addition, the endocochlear potential builds up but fails to be maintained in these mice, probably as a consequence of the loss of tight junctions between hair cells and their supporting cells [38]. The connexin 30 gene (CX30/GJB6) is contiguous with CX26/GJB2 on human chromosome 13 (mouse chromosome 14) and is also expressed in the cochlea [207, 65]. Deletions in GJB6 have been reported in deaf patients [122, 48, 156, 47]. Observations made from the first Gjb6 knock-out mouse model led to the mistaken conclusion that inactivation of Gjb6 alone could lead to deafness [207]. In fact, inactivation of the Gjb6 gene, both in humans and in mice, also impaired the expression of the Gjb2 gene [40, 174, 155, 126], and transgenic expression of Gjb2 in the same Gjb6 knock-out mouse model restored hearing [4]. Indeed, auditory brainstem responses were normal in a second, more recent Gjb6 knock-out mouse mutant, in which sufficient expression of Gjb2 was preserved. Thus, the cause of deafness after GJB6 deletion is the low expression of GJB2 due to the co-deletion of its putative regulatory element [39, 32]. In addition, the endocochlear potential in the first Gjb6 knock-out mouse model [207] fails to build up as a consequence of abnormal tight junctions between endothelial cells in capillaries of the stria vascularis [32] indicating a role of Gjb2 at this emplacement. At least, three other genes are thought to be involved in the recycling circuit of K+ ions: KCNQ4 [112], KCC3 [28], and KCC4 [27]. KCNQ4 encodes a K+ channel subunit and KCC3 and KCC4 encode K+–Cl− cotransporters. Kcnq4 is located at the base of mature OHCs and mediates a voltage-activated K+ current that is already active at the resting membrane potential [84, 103]. In Kcnq4 −/− mice, this current is abolished, leading to a slow degeneration of OHCs, which probably results from their chronic depolarisation [102]. Kcc3 and Kcc4 are present in the supporting cells of IHCs and OHCs. Kcc3 and Kcc4 are thought to siphon K+ ions from the hair cells’ pericellular space into supporting cells, where these ions would enter the gap junction recycling pathway. Hair cells undergo degeneration both in Kcc3 knock-out mice and Kcc4 knock-out mice, although degeneration occurs earlier in the former than in the latter [27, 28].
The maintenance of the high endolymphatic K+ concentration and of the endocochlear potential requires strong apical cell–cell junctions in the epithelia lining the endolymphatic compartment of the cochlea, especially in the mechanically stressed sensory epithelium. Junctions between OHCs and their supporting cells are probably subjected to the highest amount of mechanical stress, due to the motion of the sensory epithelium and forces generated by OHC electromotility. These junctions are composed of an atypical combination of tight junctions and adherens junctions [152] containing claudin-14, claudin-9, claudin-6, catenins, ZO-1, TJP2 and vezatin [22, 147, 17, 220] (see Table 1 for deafness genes). This atypical junction complex probably plays a major role in the resilience of these cell junctions to mechanical stress. Indeed, conditional mutant mice deficient for vezatin in OHCs suffer from late onset hearing loss that can also be induced irreversibly by exposure to loud sound levels that are harmless to control mice [17].
Continuing the molecular deciphering of the MET apparatus
There has been for the past 10 years remarkable progress in the identification of proteins and protein complexes that constitute the MET machinery. However, the composition of the central element of this machinery, the MET channel, is still under debate. Various strategies to characterise the molecular identity of this channel have been hindered by the limited amount of available material, the multifunction of particular molecules in the developing and mature hair bundle and by the current inability to reconstitute the MET machinery in a controlled exogenous system (see [145] for review). Genetic studies, both in humans and in mice, circumvented the problem of the paucity of the hair cell material available. The development of new genetic tools in the mouse, such as the myosin-XV promoter-driven cre mouse that enables delayed conditional knocking-out of proteins, offers a unique opportunity to distinguish the role of a particular protein in the mature hair bundle from its possible role during development [35, 160] (Fig. 2b). Other cre knock-in lines need to be developed to offer a larger panel of genetic tools at different developmental time points and in specific hair cell types. Studies that apply the same strategy to known components of the MET machinery should clarify their respective roles in the mature hair bundle.
Most genes that have been associated with deafness appear to affect MET either directly or indirectly. It is likely that the genetic approach will continue to feed the list of molecules involved in MET. As time passes, the increasing speed and smaller cost of exome sequencing will probably compensate the lower probability of finding new disease-associated loci by genetic linkage analysis of affected families. All USH1 proteins characterised so far have been implicated in the MET machinery; therefore, we can anticipate that the last USH1 protein identified, CIB2 (USH1J), will be no exception [171].
The retina is also affected by USH. The search for new binding partners of USH1 proteins in the retina is facilitated by the abundance of photoreceptor cells and may help to find new elements of the cochlear MET machinery. Until recently, the pathogenesis of the retinitis pigmentosa observed in USH1 patients remained elusive because mouse models for USH1 genetic forms do not reproduce the retinal degeneration phenotype of humans. The study of USH1 protein distribution in the macaque retina revealed the structural origin of this discrepancy [181]. In primate photoreceptor cells, USH1 proteins are present at the interface between inner and outer segments and are also associated to calyceal processes [33], which are axially oriented microvillus-like structures that form a collar around the base of the outer segment in rod and cone photoreceptors. Strikingly, calyceal processes are absent from the photoreceptor cells of mice, which probably explains the absence of an abnormal retinal phenotype in USH1 mutant mice. Calyceal processes resemble cochlear stereocilia in many respects. USH1 proteins are present in these structures, together with other molecules of the cochlear hair bundle such as myosin IIIa, espin, and the Ca2+ pump PMCA2 (plasma membrane calcium ATPase 2), which has also been implicated in mouse and human deafness [63, 202]. Furthermore, both cadherin-23 and protocadherin-15 are located at the membrane interface between the outer segment and surrounding calyceal processes and between the base of the outer segment and the apical region of the inner segment. The USH1 protein complex may form an adhesion belt connecting the outer segment basal region to the surrounding structures. These similarities between calyceal processes and hair cell stereocilia indicate that the study of photoreceptors may provide an alternative strategy to decipher the molecular elements of the MET machinery [181].
Human genetics has uncovered numerous molecules involved in hair bundle development and function. Each of these molecules provides a starting point to decipher whole molecular complexes. Clearly, the probability of finding new genes associated with deafness in patients from newly recruited families decreases with time, and as a consequence, this approach may cease to provide new candidates at some point. Moreover, lethal mutations cannot be detected by the human genetics approach, which may make some essential components of the MET machinery difficult to identify with this approach. Thus, complementary strategies need to be developed to complete the picture of the molecular networks in the hair bundle. In addition to the yeast two-hybrid technique that can find new interacting components of a molecular complex step by step [114, 133, 237], recent technological leaps have offered new screening strategies. Analysis of isolated hair bundles by mass spectroscopy could establish an extensive list of hair bundle proteins and their relative abundances, which would provide a new framework to pursue functional studies. Among the most abundant proteins, many are involved in the organisation of the actin cytoskeleton, in the maintenance of local ATP levels (the brain isoform of creatine kinase) [197, 196, 12], in calcium homeostasis (calcium buffering proteins such as parvalbumin, calbindin and calmodulin [80, 197], and the Ca2+ pump PMCA2 [63, 202]). Likewise, next generation sequencing coupled with messenger RNA amplification of a few sensory hair cells should bring new insight into the molecular components involved in hair cell MET. The variety of structures in which these components are involved implies that the understanding of their functions will rely more and more on in vivo studies in the future. Genetically modified mice have proven to be a powerful tool to study the role of molecules in situ. In addition, the replication of relevant human point mutations in mice has been very instructive, as illustrated by the use of particular Tecta and Tectb mutations to uncover the various roles of the tectorial membrane in MET. This mutational approach is to be extended with the arrival of more powerful and faster tools to engineer mouse mutants, such as the clustered regularly interspaced short palindromic repeat/CRISPR-associated (CRISPR/Cas) system to perform genome sequence specific-editing. The CRISPR/Cas system allows the one-step generation of mice carrying mutations in several genes simultaneously [224]. This system also offers the possibility to generate reporter and conditional alleles in one step [241], and hence speeds up considerably the generation of genetic models in mice. This gene editing method has already been applied to zebrafish [36, 88], and should also make it possible to manipulate the genomes of other mammalian species, including ones that have a frequency range of hearing more similar to that of humans, such as guinea pig or gerbil.
Abbreviations
- Alms1:
-
Alström syndrome 1
- aPKC:
-
Atypical protein kinase C
- BBS1/4:
-
Bardet–Biedl syndrome 1/4
- CEACAM16:
-
Carcinoembryonic antigen-related cell adhesion molecule 16
- cKO:
-
Conditional knock-out
- CLIC5:
-
Cl- intracellular channel 5
- CRISPR/Cas:
-
Clustered regularly interspaced short palindromic repeat/CRISPR-associated
- CTHRC1:
-
Collagen triple helix repeat containing 1
- cx26:
-
Connexin 26
- cx30:
-
Connexin 30
- dchs1:
-
Dachsous 1
- ELMO:
-
Engulfment and cell motility
- ERM:
-
Ezrin/radixin/moesin
- eps8:
-
Epidermal growth factor receptor pathway substrate 8
- Fat4:
-
FAT tumor suppressor homolog 4
- Gαi :
-
GTP-binding protein alpha-i subunit
- GFP:
-
Green fluorescent protein
- GJB2:
-
Gap junction protein beta 2 (connexin 26)
- GJB6:
-
Gap junction protein beta 6 (connexin 30)
- GPSM2:
-
G-protein signaling modulator 2
- ift88:
-
Intraflagellar transport 88 homolog
- IHC:
-
Inner hair cell
- KCC3/4:
-
K+/Cl- cotransporter 3/4
- Kcne1:
-
K+ voltage-gated channel, Isk-related subfamily, member 1
- Kcnj10:
-
K+ inwardly rectifying channel, subfamily J, member 10
- Kcnq1/4:
-
K+ voltage-gated channel, subfamily Q, member 1/4
- kif3a:
-
Kinesin family member 3A
- KO:
-
Knock-out
- LOXHD1:
-
Lipoxygenase homology domains 1
- MAGI1:
-
Membrane-associated guanylate kinase inverted 1
- MAGUK:
-
Membrane-associated guanylate kinase
- MET:
-
Mechano-electrical transduction
- MKKS:
-
McKusick–Kaufman syndrome
- Mks1:
-
Meckel syndrome, type 1
- NHERF1/2:
-
Na+/H+ exchanger regulatory factor 1/2
- NKCC1:
-
Na+–K+–2Cl− cotransporter
- OHC:
-
Outer hair cell
- PDZ:
-
Postsynaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1) and zonula occludens-1 protein (ZO-1)
- PDZD7:
-
PDZ domain containing 7
- PMCA2:
-
Plasma membrane Ca2+ ATPase 2
- PST:
-
Proline-serine-threonine rich domain
- PTK7:
-
Protein tyrosine kinase 7
- PTPRQ:
-
Protein tyrosine phosphatase receptor Q
- rdx:
-
Radixin
- ror2:
-
Receptor tyrosine kinase-like orphan receptor 2
- scrib:
-
Scribbled
- sec24b:
-
Sec24 family member B
- smurf1/2:
-
SMAD-specific E3 ubiquitin protein ligase 1/2
- TJP2:
-
Tight junction protein 2
- TRIOBP:
-
TRIO and F-actin binding protein
- TTC8:
-
Tetratricopeptide repeat domain 8
- USH:
-
Usher syndrome
- vangl1/2:
-
vang-like 1/2
- VLGR1:
-
Very large G-coupled receptor 1
- ZO-1:
-
Zonula occludens 1
References
Adato A, Lefevre G, Delprat B, Michel V, Michalski N, Chardenoux S, Weil D, El-Amraoui A, Petit C (2005) Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum Mol Genet 14(24):3921–3932. doi:10.1093/hmg/ddi416
Adato A, Michel V, Kikkawa Y, Reiners J, Alagramam KN, Weil D, Yonekawa H, Wolfrum U, El-Amraoui A, Petit C (2005) Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet 14(3):347–356. doi:10.1093/hmg/ddi031
Adato A, Vreugde S, Joensuu T, Avidan N, Hamalainen R, Belenkiy O, Olender T, Bonne-Tamir B, Ben-Asher E, Espinos C, Millan JM, Lehesjoki AE, Flannery JG, Avraham KB, Pietrokovski S, Sankila EM, Beckmann JS, Lancet D (2002) USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur J Hum Genet 10(6):339–350. doi:10.1038/sj.ejhg.5200831
Ahmad S, Tang W, Chang Q, Qu Y, Hibshman J, Li Y, Sohl G, Willecke K, Chen P, Lin X (2007) Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human connexin30-linked deafness. Proc Natl Acad Sci U S A 104(4):1337–1341. doi:10.1073/pnas.0606855104
Ahmed ZM, Goodyear R, Riazuddin S, Lagziel A, Legan PK, Behra M, Burgess SM, Lilley KS, Wilcox ER, Griffith AJ, Frolenkov GI, Belyantseva IA, Richardson GP, Friedman TB (2006) The tip-link antigen, a protein associated with the transduction complex of sensory hair cells, is protocadherin-15. J Neurosci 26(26):7022–7034. doi:10.1523/JNEUROSCI.1163-06.2006
Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, Morell RJ, Friedman TB, Wilcox ER (2001) Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet 69(1):25–34. doi:10.1086/321277
Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP (2001) The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat Genet 27(1):99–102. doi:10.1038/83837
Alagramam KN, Yuan H, Kuehn MH, Murcia CL, Wayne S, Srisailpathy CR, Lowry RB, Knaus R, Van Laer L, Bernier FP, Schwartz S, Lee C, Morton CC, Mullins RF, Ramesh A, Van Camp G, Hageman GS, Woychik RP, Smith RJ (2001) Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum Mol Genet 10(16):1709–1718
Assad JA, Corey DP (1992) An active motor model for adaptation by vertebrate hair cells. J Neurosci 12(9):3291–3309
Assad JA, Shepherd GM, Corey DP (1991) Tip-link integrity and mechanical transduction in vertebrate hair cells. Neuron 7(6):985–994
Avan P, Buki B, Petit C (2013) Auditory distortions: origins and functions. Physiol Rev 93(4):1563–1619. doi:10.1152/physrev.00029.2012
Avenarius MR, Saylor KW, Lundeberg MR, Wilmarth PA, Shin JB, Spinelli KJ, Pagana JM, Andrade L, Kachar B, Choi D, David LL, Barr-Gillespie PG (2013) Correlation of actin crosslinker and capper expression levels with stereocilia growth phases. Mol Cell Proteomics. doi:10.1074/mcp.M113.033704
Avraham KB, Hasson T, Sobe T, Balsara B, Testa JR, Skvorak AB, Morton CC, Copeland NG, Jenkins NA (1997) Characterization of unconventional MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice. Hum Mol Genet 6(8):1225–1231
Avraham KB, Hasson T, Steel KP, Kingsley DM, Russell LB, Mooseker MS, Copeland NG, Jenkins NA (1995) The mouse Snell’s waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nat Genet 11(4):369–375. doi:10.1038/ng1295-369
Azaiez H, Booth KT, Bu F, Huygen P, Shibata SB, Shearer AE, Kolbe D, Meyer N, Black-Ziegelbein EA, Smith RJ (2014) TBC1D24 mutation causes autosomal-dominant nonsyndromic hearing loss. Hum Mutat. doi:10.1002/humu.22557
Bahloul A, Michel V, Hardelin JP, Nouaille S, Hoos S, Houdusse A, England P, Petit C (2010) Cadherin-23, myosin VIIa and harmonin, encoded by Usher syndrome type I genes, form a ternary complex and interact with membrane phospholipids. Hum Mol Genet 19(18):3557–3565. doi:10.1093/hmg/ddq271
Bahloul A, Simmler MC, Michel V, Leibovici M, Perfettini I, Roux I, Weil D, Nouaille S, Zuo J, Zadro C, Licastro D, Gasparini P, Avan P, Hardelin JP, Petit C (2009) Vezatin, an integral membrane protein of adherens junctions, is required for the sound resilience of cochlear hair cells. EMBO Mol Med 1(2):125–138. doi:10.1002/emmm.200900015
Barr-Gillespie PG, Nicolson T (2013) Who needs tip links? Backwards transduction by hair cells. J Gen Physiol 142(5):481–486. doi:10.1085/jgp.201311111
Bashtanov ME, Goodyear RJ, Richardson GP, Russell IJ (2004) The mechanical properties of chick (Gallus domesticus) sensory hair bundles: relative contributions of structures sensitive to calcium chelation and subtilisin treatment. J Physiol 559(Pt 1):287–299. doi:10.1113/jphysiol.2004.065565
Behlouli A, Bonnet C, Abdi S, Bouaita A, Lelli A, Hardelin JP, Schietroma C, Rous Y, Louha M, Cheknane A, Lebdi H, Boudjelida K, Makrelouf M, Zenati A, Petit C (2014) EPS8, encoding an actin-binding protein of cochlear hair cell stereocilia, is a new causal gene for autosomal recessive profound deafness. Orphanet J Rare Dis 9(1):55. doi:10.1186/1750-1172-9-55
Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, Ahmed ZM, Griffith AJ, Friedman TB (2005) Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol 7(2):148–156. doi:10.1038/ncb1219
Ben-Yosef T, Belyantseva IA, Saunders TL, Hughes ED, Kawamoto K, Van Itallie CM, Beyer LA, Halsey K, Gardner DJ, Wilcox ER, Rasmussen J, Anderson JM, Dolan DF, Forge A, Raphael Y, Camper SA, Friedman TB (2003) Claudin 14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum Mol Genet 12(16):2049–2061
Beurg M, Evans MG, Hackney CM, Fettiplace R (2006) A large-conductance calcium-selective mechanotransducer channel in mammalian cochlear hair cells. J Neurosci 26(43):10992–11000. doi:10.1523/JNEUROSCI.2188-06.2006
Beurg M, Fettiplace R, Nam JH, Ricci AJ (2009) Localization of inner hair cell mechanotransducer channels using high-speed calcium imaging. Nat Neurosci 12(5):553–558. doi:10.1038/nn.2295
Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, Hussain K, Furth-Lavi J, Cosgrove KE, Shepherd RM, Barnes PD, O’Brien RE, Farndon PA, Sowden J, Liu XZ, Scanlan MJ, Malcolm S, Dunne MJ, Aynsley-Green A, Glaser B (2000) A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat Genet 26(1):56–60. doi:10.1038/79178
Boeda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners J, Houdusse A, Legrain P, Wolfrum U, Richardson G, Petit C (2002) Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J 21(24):6689–6699
Boettger T, Hubner CA, Maier H, Rust MB, Beck FX, Jentsch TJ (2002) Deafness and renal tubular acidosis in mice lacking the K–Cl co-transporter Kcc4. Nature 416(6883):874–878. doi:10.1038/416874a
Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, Lemcke B, Horst J, Leuwer R, Pape HC, Volkl H, Hubner CA, Jentsch TJ (2003) Loss of K–Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J 22(20):5422–5434. doi:10.1093/emboj/cdg519
Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG, Seeliger M, C-Salcedó Cabrera M, Vila MC, Molina OP, Gal A, Kubisch C (2001) Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet 27(1):108–112. doi:10.1038/83667
Bonnet C, Louha M, Loundon N, Michalski N, Verpy E, Smagghe L, Hardelin JP, Rouillon I, Jonard L, Couderc R, Gherbi S, Garabedian EN, Denoyelle F, Petit C, Marlin S (2013) Biallelic nonsense mutations in the otogelin-like gene (OTOGL) in a child affected by mild to moderate hearing impairment. Gene 527(2):537–540. doi:10.1016/j.gene.2013.06.044
Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR, Wayne S, Bellman S, Desmukh D, Ahmed Z, Khan SN, Kaloustian VM, Li XC, Lalwani A, Bitner-Glindzicz M, Nance WE, Liu XZ, Wistow G, Smith RJ, Griffith AJ, Wilcox ER, Friedman TB, Morell RJ (2001) Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet 68(1):26–37. doi:10.1086/316954
Boulay AC, del Castillo FJ, Giraudet F, Hamard G, Giaume C, Petit C, Avan P, Cohen-Salmon M (2013) Hearing is normal without connexin30. J Neurosci 33(2):430–434. doi:10.1523/JNEUROSCI.4240-12.2013
Brown PK, Gibbons IR, Wald G (1963) The visual cells and visual pigment of the mudpuppy, necturus. J Cell Biol 19:79–106
Caberlotto E, Michel V, de Monvel JB, Petit C (2011) Coupling of the mechanotransduction machinery and F-actin polymerization in the cochlear hair bundles. Bioarchitecture 1(4):169–174. doi:10.4161/bioa.1.4.17532
Caberlotto E, Michel V, Foucher I, Bahloul A, Goodyear RJ, Pepermans E, Michalski N, Perfettini I, Alegria-Prevot O, Chardenoux S, Do Cruzeiro M, Hardelin JP, Richardson GP, Avan P, Weil D, Petit C (2011) Usher type 1G protein sans is a critical component of the tip-link complex, a structure controlling actin polymerization in stereocilia. Proc Natl Acad Sci U S A 108(14):5825–5830. doi:10.1073/pnas.1017114108
Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, Xiong JW, Xi JJ (2013) Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res 23(4):465–472. doi:10.1038/cr.2013.45
Cohen-Salmon M, El-Amraoui A, Leibovici M, Petit C (1997) Otogelin: a glycoprotein specific to the acellular membranes of the inner ear. Proc Natl Acad Sci U S A 94(26):14450–14455
Cohen-Salmon M, Ott T, Michel V, Hardelin JP, Perfettini I, Eybalin M, Wu T, Marcus DC, Wangemann P, Willecke K, Petit C (2002) Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr Biol 12(13):1106–1111
Cohen-Salmon M, Regnault B, Cayet N, Caille D, Demuth K, Hardelin JP, Janel N, Meda P, Petit C (2007) Connexin30 deficiency causes instrastrial fluid-blood barrier disruption within the cochlear stria vascularis. Proc Natl Acad Sci U S A 104(15):6229–6234. doi:10.1073/pnas.0605108104
Common JE, Bitner-Glindzicz M, O’Toole EA, Barnes MR, Jenkins L, Forge A, Kelsell DP (2005) Specific loss of connexin 26 expression in ductal sweat gland epithelium associated with the deletion mutation del(GJB6-D13S1830). Clin Exp Dermatol 30(6):688–693. doi:10.1111/j.1365-2230.2005.01878.x
Corey DP, Hudspeth AJ (1979) Ionic basis of the receptor potential in a vertebrate hair cell. Nature 281(5733):675–677
Corey DP, Hudspeth AJ (1983) Kinetics of the receptor current in bullfrog saccular hair cells. J Neurosci 3(5):962–976
Crawford AC, Evans MG, Fettiplace R (1991) The actions of calcium on the mechano-electrical transducer current of turtle hair cells. J Physiol 434:369–398
Cui C, Chatterjee B, Francis D, Yu Q, SanAgustin JT, Francis R, Tansey T, Henry C, Wang B, Lemley B, Pazour GJ, Lo CW (2011) Disruption of Mks1 localization to the mother centriole causes cilia defects and developmental malformations in Meckel–Gruber syndrome. Dis Model Mech 4(1):43–56. doi:10.1242/dmm.006262
Dallos P (1985) Response characteristics of mammalian cochlear hair cells. J Neurosci 5(6):1591–1608
Deans MR, Peterson JM, Wong GW (2010) Mammalian Otolin: a multimeric glycoprotein specific to the inner ear that interacts with otoconial matrix protein Otoconin-90 and Cerebellin-1. PLoS One 5(9):e12765. doi:10.1371/journal.pone.0012765
del Castillo FJ, Rodriguez-Ballesteros M, Alvarez A, Hutchin T, Leonardi E, de Oliveira CA, Azaiez H, Brownstein Z, Avenarius MR, Marlin S, Pandya A, Shahin H, Siemering KR, Weil D, Wuyts W, Aguirre LA, Martin Y, Moreno-Pelayo MA, Villamar M, Avraham KB, Dahl HH, Kanaan M, Nance WE, Petit C, Smith RJ, Van Camp G, Sartorato EL, Murgia A, Moreno F, del Castillo I (2005) A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J Med Genet 42(7):588–594. doi:10.1136/jmg.2004.028324
del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, Menendez I, Moreno F (2002) A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 346(4):243–249. doi:10.1056/NEJMoa012052
Delpire E, Lu J, England R, Dull C, Thorne T (1999) Deafness and imbalance associated with inactivation of the secretory Na–K–2Cl co-transporter. Nat Genet 22(2):192–195. doi:10.1038/9713
Delprat B, Michel V, Goodyear R, Yamasaki Y, Michalski N, El-Amraoui A, Perfettini I, Legrain P, Richardson G, Hardelin JP, Petit C (2005) Myosin XVa and whirlin, two deafness gene products required for hair bundle growth, are located at the stereocilia tips and interact directly. Hum Mol Genet 14(3):401–410. doi:10.1093/hmg/ddi036
Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K (2001) Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet 27(1):103–107. doi:10.1038/83660
Dixon MJ, Gazzard J, Chaudhry SS, Sampson N, Schulte BA, Steel KP (1999) Mutation of the Na–K–Cl co-transporter gene Slc12a2 results in deafness in mice. Hum Mol Genet 8(8):1579–1584
Doherty D, Chudley AE, Coghlan G, Ishak GE, Innes AM, Lemire EG, Rogers RC, Mhanni AA, Phelps IG, Jones SJ, Zhan SH, Fejes AP, Shahin H, Kanaan M, Akay H, Tekin M, Triggs-Raine B, Zelinski T (2012) GPSM2 mutations cause the brain malformations and hearing loss in Chudley–McCullough syndrome. Am J Hum Genet 90(6):1088–1093. doi:10.1016/j.ajhg.2012.04.008
Donowitz M, Cha B, Zachos NC, Brett CL, Sharma A, Tse CM, Li X (2005) NHERF family and NHE3 regulation. J Physiol 567(Pt 1):3–11. doi:10.1113/jphysiol.2005.090399
Eatock RA (2000) Adaptation in hair cells. Annu Rev Neurosci 23:285–314. doi:10.1146/annurev.neuro.23.1.285
Ebermann I, Phillips JB, Liebau MC, Koenekoop RK, Schermer B, Lopez I, Schafer E, Roux AF, Dafinger C, Bernd A, Zrenner E, Claustres M, Blanco B, Nurnberg G, Nurnberg P, Ruland R, Westerfield M, Benzing T, Bolz HJ (2010) PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J Clin Invest 120(6):1812–1823. doi:10.1172/JCI39715
Ebermann I, Scholl HP, Charbel Issa P, Becirovic E, Lamprecht J, Jurklies B, Millan JM, Aller E, Mitter D, Bolz H (2007) A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet 121(2):203–211. doi:10.1007/s00439-006-0304-0
Etheridge SL, Ray S, Li S, Hamblet NS, Lijam N, Tsang M, Greer J, Kardos N, Wang J, Sussman DJ, Chen P, Wynshaw-Boris A (2008) Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet 4(11):e1000259. doi:10.1371/journal.pgen.1000259
Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, Yan D, Ahmad I, Cheng JJ, Ayuso C, Cremers C, Davenport S, Moller C, Talmadge CB, Beisel KW, Tamayo M, Morton CC, Swaroop A, Kimberling WJ, Sumegi J (1998) Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science 280(5370):1753–1757
Ezan J, Lasvaux L, Gezer A, Novakovic A, May-Simera H, Belotti E, Lhoumeau AC, Birnbaumer L, Beer-Hammer S, Borg JP, Le Bivic A, Nurnberg B, Sans N, Montcouquiol M (2013) Primary cilium migration depends on G-protein signalling control of subapical cytoskeleton. Nat Cell Biol 15(9):1107–1115. doi:10.1038/ncb2819
Ezan J, Montcouquiol M (2013) Revisiting planar cell polarity in the inner ear. Semin Cell Dev Biol 24(5):499–506. doi:10.1016/j.semcdb.2013.03.012
Farris HE, LeBlanc CL, Goswami J, Ricci AJ (2004) Probing the pore of the auditory hair cell mechanotransducer channel in turtle. J Physiol 558(Pt 3):769–792. doi:10.1113/jphysiol.2004.061267
Ficarella R, Di Leva F, Bortolozzi M, Ortolano S, Donaudy F, Petrillo M, Melchionda S, Lelli A, Domi T, Fedrizzi L, Lim D, Shull GE, Gasparini P, Brini M, Mammano F, Carafoli E (2007) A functional study of plasma-membrane calcium-pump isoform 2 mutants causing digenic deafness. Proc Natl Acad Sci U S A 104(5):1516–1521. doi:10.1073/pnas.0609775104
Fields RR, Zhou G, Huang D, Davis JR, Moller C, Jacobson SG, Kimberling WJ, Sumegi J (2002) Usher syndrome type III: revised genomic structure of the USH3 gene and identification of novel mutations. Am J Hum Genet 71(3):607–617. doi:10.1086/342098
Forge A, Jagger DJ, Kelly JJ, Taylor RR (2013) Connexin30-mediated intercellular communication plays an essential role in epithelial repair in the cochlea. J Cell Sci 126(Pt 7):1703–1712. doi:10.1242/jcs.125476
Forsythe E, Beales PL (2013) Bardet–Biedl syndrome. Eur J Hum Genet 21(1):8–13. doi:10.1038/ejhg.2012.115
Furness DN, Hackney CM (1985) Cross-links between stereocilia in the guinea pig cochlea. Hear Res 18(2):177–188
Furness DN, Johnson SL, Manor U, Ruttiger L, Tocchetti A, Offenhauser N, Olt J, Goodyear RJ, Vijayakumar S, Dai Y, Hackney CM, Franz C, Di Fiore PP, Masetto S, Jones SM, Knipper M, Holley MC, Richardson GP, Kachar B, Marcotti W (2013) Progressive hearing loss and gradual deterioration of sensory hair bundles in the ears of mice lacking the actin-binding protein Eps8L2. Proc Natl Acad Sci U S A 110(34):13898–13903. doi:10.1073/pnas.1304644110
Furness DN, Mahendrasingam S, Ohashi M, Fettiplace R, Hackney CM (2008) The dimensions and composition of stereociliary rootlets in mammalian cochlear hair cells: comparison between high- and low-frequency cells and evidence for a connection to the lateral membrane. J Neurosci 28(25):6342–6353. doi:10.1523/JNEUROSCI.1154-08.2008
Gagnon LH, Longo-Guess CM, Berryman M, Shin JB, Saylor KW, Yu H, Gillespie PG, Johnson KR (2006) The chloride intracellular channel protein CLIC5 is expressed at high levels in hair cell stereocilia and is essential for normal inner ear function. J Neurosci 26(40):10188–10198. doi:10.1523/JNEUROSCI.2166-06.2006
Geleoc GS, Lennan GW, Richardson GP, Kros CJ (1997) A quantitative comparison of mechanoelectrical transduction in vestibular and auditory hair cells of neonatal mice. Proc Biol Sci 264(1381):611–621. doi:10.1098/rspb.1997.0087
Gibson F, Walsh J, Mburu P, Varela A, Brown KA, Antonio M, Beisel KW, Steel KP, Brown SD (1995) A type VII myosin encoded by the mouse deafness gene shaker-1. Nature 374(6517):62–64. doi:10.1038/374062a0
Giese AP, Ezan J, Wang L, Lasvaux L, Lembo F, Mazzocco C, Richard E, Reboul J, Borg JP, Kelley MW, Sans N, Brigande J, Montcouquiol M (2012) Gipc1 has a dual role in Vangl2 trafficking and hair bundle integrity in the inner ear. Development 139(20):3775–3785. doi:10.1242/dev.074229
Goodyear RJ, Legan PK, Wright MB, Marcotti W, Oganesian A, Coats SA, Booth CJ, Kros CJ, Seifert RA, Bowen-Pope DF, Richardson GP (2003) A receptor-like inositol lipid phosphatase is required for the maturation of developing cochlear hair bundles. J Neurosci 23(27):9208–9219
Goodyear RJ, Marcotti W, Kros CJ, Richardson GP (2005) Development and properties of stereociliary link types in hair cells of the mouse cochlea. J Comp Neurol 485(1):75–85. doi:10.1002/cne.20513
Grati M, Kachar B (2011) Myosin VIIa and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc Natl Acad Sci U S A 108(28):11476–11481. doi:10.1073/pnas.1104161108
Grati M, Shin JB, Weston MD, Green J, Bhat MA, Gillespie PG, Kachar B (2012) Localization of PDZD7 to the stereocilia ankle-link associates this scaffolding protein with the Usher syndrome protein network. J Neurosci 32(41):14288–14293. doi:10.1523/JNEUROSCI.3071-12.2012
Grillet N, Schwander M, Hildebrand MS, Sczaniecka A, Kolatkar A, Velasco J, Webster JA, Kahrizi K, Najmabadi H, Kimberling WJ, Stephan D, Bahlo M, Wiltshire T, Tarantino LM, Kuhn P, Smith RJ, Muller U (2009) Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am J Hum Genet 85(3):328–337. doi:10.1016/j.ajhg.2009.07.017
Grillet N, Xiong W, Reynolds A, Kazmierczak P, Sato T, Lillo C, Dumont RA, Hintermann E, Sczaniecka A, Schwander M, Williams D, Kachar B, Gillespie PG, Muller U (2009) Harmonin mutations cause mechanotransduction defects in cochlear hair cells. Neuron 62(3):375–387. doi:10.1016/j.neuron.2009.04.006
Hackney CM, Mahendrasingam S, Penn A, Fettiplace R (2005) The concentrations of calcium buffering proteins in mammalian cochlear hair cells. J Neurosci 25(34):7867–7875. doi:10.1523/JNEUROSCI.1196-05.2005
Hofer D, Ness W, Drenckhahn D (1997) Sorting of actin isoforms in chicken auditory hair cells. J Cell Sci 110(Pt 6):765–770
Holt JR, Gillespie SK, Provance DW, Shah K, Shokat KM, Corey DP, Mercer JA, Gillespie PG (2002) A chemical-genetic strategy implicates myosin-1c in adaptation by hair cells. Cell 108(3):371–381
Holt JR, Pan B, Koussa MA, Asai Y (2014) TMC function in hair cell transduction. Hear Res. doi:10.1016/j.heares.2014.01.001
Housley GD, Ashmore JF (1992) Ionic currents of outer hair cells isolated from the guinea-pig cochlea. J Physiol 448:73–98
Howard J, Hudspeth AJ (1987) Mechanical relaxation of the hair bundle mediates adaptation in mechanoelectrical transduction by the bullfrog’s saccular hair cell. Proc Natl Acad Sci U S A 84(9):3064–3068
Hudspeth AJ (1982) Extracellular current flow and the site of transduction by vertebrate hair cells. J Neurosci 2(1):1–10
Hudspeth AJ, Corey DP (1977) Sensitivity, polarity, and conductance change in the response of vertebrate hair cells to controlled mechanical stimuli. Proc Natl Acad Sci U S A 74(6):2407–2411
Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, Joung JK (2013) Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31(3):227–229. doi:10.1038/nbt.2501
Jagger D, Collin G, Kelly J, Towers E, Nevill G, Longo-Guess C, Benson J, Halsey K, Dolan D, Marshall J, Naggert J, Forge A (2011) Alstrom syndrome protein ALMS1 localizes to basal bodies of cochlear hair cells and regulates cilium-dependent planar cell polarity. Hum Mol Genet 20(3):466–481. doi:10.1093/hmg/ddq493
Jaworek TJ, Richard EM, Ivanova AA, Giese AP, Choo DI, Khan SN, Riazuddin S, Kahn RA (2013) An alteration in ELMOD3, an Arl2 GTPase-activating protein, is associated with hearing impairment in humans. PLoS Genet 9(9):e1003774. doi:10.1371/journal.pgen.1003774
Joensuu T, Hamalainen R, Yuan B, Johnson C, Tegelberg S, Gasparini P, Zelante L, Pirvola U, Pakarinen L, Lehesjoki AE, de la Chapelle A, Sankila EM (2001) Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet 69(4):673–684. doi:10.1086/323610
Johnson KR, Gagnon LH, Webb LS, Peters LL, Hawes NL, Chang B, Zheng QY (2003) Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum Mol Genet 12(23):3075–3086. doi:10.1093/hmg/ddg332
Johnson KR, Longo-Guess CM, Gagnon LH (2012) Mutations of the mouse ELMO domain containing 1 gene (Elmod1) link small GTPase signaling to actin cytoskeleton dynamics in hair cell stereocilia. PLoS One 7(4):e36074. doi:10.1371/journal.pone.0036074
Johnson SL, Beurg M, Marcotti W, Fettiplace R (2011) Prestin-driven cochlear amplification is not limited by the outer hair cell membrane time constant. Neuron 70(6):1143–1154. doi:10.1016/j.neuron.2011.04.024
Jones C, Roper VC, Foucher I, Qian D, Banizs B, Petit C, Yoder BK, Chen P (2008) Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet 40(1):69–77. doi:10.1038/ng.2007.54
Kamiya K, Michel V, Giraudet F, Riederer B, Foucher I, Papal S, Perfettini I, Le Gal S, Verpy E, Xia W, Seidler U, Georgescu MM, Avan P, El-Amraoui A, Petit C (2014) An unusually powerful mode of low-frequency sound interference due to defective hair bundles of the auditory outer hair cells. Proc Natl Acad Sci U S A. doi:10.1073/pnas1405322111
Kammerer R, Ruttiger L, Riesenberg R, Schauble C, Krupar R, Kamp A, Sunami K, Eisenried A, Hennenberg M, Grunert F, Bress A, Battaglia S, Schrewe H, Knipper M, Schneider MR, Zimmermann W (2012) Loss of mammal-specific tectorial membrane component carcinoembryonic antigen cell adhesion molecule 16 (CEACAM16) leads to hearing impairment at low and high frequencies. J Biol Chem 287(26):21584–21598. doi:10.1074/jbc.M111.320481
Kawashima Y, Geleoc GS, Kurima K, Labay V, Lelli A, Asai Y, Makishima T, Wu DK, Della Santina CC, Holt JR, Griffith AJ (2011) Mechanotransduction in mouse inner ear hair cells requires transmembrane channel-like genes. J Clin Invest 121(12):4796–4809. doi:10.1172/JCI60405
Kazmierczak P, Sakaguchi H, Tokita J, Wilson-Kubalek EM, Milligan RA, Muller U, Kachar B (2007) Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature 449(7158):87–91. doi:10.1038/nature06091
Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM (1997) Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 387(6628):80–83. doi:10.1038/387080a0
Khan SY, Ahmed ZM, Shabbir MI, Kitajiri S, Kalsoom S, Tasneem S, Shayiq S, Ramesh A, Srisailpathy S, Khan SN, Smith RJ, Riazuddin S, Friedman TB (2007) Mutations of the RDX gene cause nonsyndromic hearing loss at the DFNB24 locus. Hum Mutat 28(5):417–423. doi:10.1002/humu.20469
Kharkovets T, Dedek K, Maier H, Schweizer M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T, Jentsch TJ (2006) Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J 25(3):642–652. doi:10.1038/sj.emboj.7600951
Kharkovets T, Hardelin JP, Safieddine S, Schweizer M, El-Amraoui A, Petit C, Jentsch TJ (2000) KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci U S A 97(8):4333–4338
Kikkawa Y, Shitara H, Wakana S, Kohara Y, Takada T, Okamoto M, Taya C, Kamiya K, Yoshikawa Y, Tokano H, Kitamura K, Shimizu K, Wakabayashi Y, Shiroishi T, Kominami R, Yonekawa H (2003) Mutations in a new scaffold protein Sans cause deafness in Jackson shaker mice. Hum Mol Genet 12(5):453–461
Kim KX, Beurg M, Hackney CM, Furness DN, Mahendrasingam S, Fettiplace R (2013) The role of transmembrane channel-like proteins in the operation of hair cell mechanotransducer channels. J Gen Physiol 142(5):493–505. doi:10.1085/jgp.201311068
Kim KX, Fettiplace R (2013) Developmental changes in the cochlear hair cell mechanotransducer channel and their regulation by transmembrane channel-like proteins. J Gen Physiol 141(1):141–148. doi:10.1085/jgp.201210913
Kitajiri S, Fukumoto K, Hata M, Sasaki H, Katsuno T, Nakagawa T, Ito J, Tsukita S (2004) Radixin deficiency causes deafness associated with progressive degeneration of cochlear stereocilia. J Cell Biol 166(4):559–570. doi:10.1083/jcb.200402007
Kitajiri S, Sakamoto T, Belyantseva IA, Goodyear RJ, Stepanyan R, Fujiwara I, Bird JE, Riazuddin S, Ahmed ZM, Hinshaw JE, Sellers J, Bartles JR, Hammer JA 3rd, Richardson GP, Griffith AJ, Frolenkov GI, Friedman TB (2010) Actin-bundling protein TRIOBP forms resilient rootlets of hair cell stereocilia essential for hearing. Cell 141(5):786–798. doi:10.1016/j.cell.2010.03.049
Kitamura K, Kakoi H, Yoshikawa Y, Ochikubo F (1992) Ultrastructural findings in the inner ear of Jackson shaker mice. Acta Otolaryngol 112(4):622–627
Kros CJ, Marcotti W, van Netten SM, Self TJ, Libby RT, Brown SD, Richardson GP, Steel KP (2002) Reduced climbing and increased slipping adaptation in cochlear hair cells of mice with Myo7a mutations. Nat Neurosci 5(1):41–47. doi:10.1038/nn784
Kros CJ, Rusch A, Richardson GP (1992) Mechano-electrical transducer currents in hair cells of the cultured neonatal mouse cochlea. Proc Biol Sci 249(1325):185–193. doi:10.1098/rspb.1992.0102
Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ (1999) KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96(3):437–446
Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, Arnaud D, Drury S, Mo J, Makishima T, Ghosh M, Menon PS, Deshmukh D, Oddoux C, Ostrer H, Khan S, Deininger PL, Hampton LL, Sullivan SL, Battey JF Jr, Keats BJ, Wilcox ER, Friedman TB, Griffith AJ (2002) Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet 30(3):277–284. doi:10.1038/ng842
Kussel-Andermann P, El-Amraoui A, Safieddine S, Nouaille S, Perfettini I, Lecuit M, Cossart P, Wolfrum U, Petit C (2000) Vezatin, a novel transmembrane protein, bridges myosin VIIA to the cadherin-catenins complex. EMBO J 19(22):6020–6029. doi:10.1093/emboj/19.22.6020
Lee J, Andreeva A, Sipe CW, Liu L, Cheng A, Lu X (2012) PTK7 regulates myosin II activity to orient planar polarity in the mammalian auditory epithelium. Curr Biol 22(11):956–966. doi:10.1016/j.cub.2012.03.068
Lee MP, Ravenel JD, Hu RJ, Lustig LR, Tomaselli G, Berger RD, Brandenburg SA, Litzi TJ, Bunton TE, Limb C, Francis H, Gorelikow M, Gu H, Washington K, Argani P, Goldenring JR, Coffey RJ, Feinberg AP (2000) Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J Clin Invest 106(12):1447–1455. doi:10.1172/JCI10897
Lefevre G, Michel V, Weil D, Lepelletier L, Bizard E, Wolfrum U, Hardelin JP, Petit C (2008) A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development 135(8):1427–1437. doi:10.1242/dev.012922
Legan PK, Goodyear RJ, Morin M, Mencia A, Pollard H, Olavarrieta L, Korchagina J, Modamio-Hoybjor S, Mayo F, Moreno F, Moreno-Pelayo MA, Richardson GP (2014) Three deaf mice: mouse models for TECTA-based human hereditary deafness reveal domain-specific structural phenotypes in the tectorial membrane. Hum Mol Genet. doi:10.1093/hmg/ddt646
Legan PK, Lukashkina VA, Goodyear RJ, Kossi M, Russell IJ, Richardson GP (2000) A targeted deletion in alpha-tectorin reveals that the tectorial membrane is required for the gain and timing of cochlear feedback. Neuron 28(1):273–285
Legan PK, Lukashkina VA, Goodyear RJ, Lukashkin AN, Verhoeven K, Van Camp G, Russell IJ, Richardson GP (2005) A deafness mutation isolates a second role for the tectorial membrane in hearing. Nat Neurosci 8(8):1035–1042. doi:10.1038/nn1496
Legan PK, Rau A, Keen JN, Richardson GP (1997) The mouse tectorins. Modular matrix proteins of the inner ear homologous to components of the sperm–egg adhesion system. J Biol Chem 272(13):8791–8801
Lerer I, Sagi M, Ben-Neriah Z, Wang T, Levi H, Abeliovich D (2001) A deletion mutation in GJB6 cooperating with a GJB2 mutation in trans in non-syndromic deafness: a novel founder mutation in Ashkenazi Jews. Hum Mutat 18(5):460. doi:10.1002/humu.1222
Lu X, Borchers AG, Jolicoeur C, Rayburn H, Baker JC, Tessier-Lavigne M (2004) PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature 430(6995):93–98. doi:10.1038/nature02677
Lukashkin AN, Legan PK, Weddell TD, Lukashkina VA, Goodyear RJ, Welstead LJ, Petit C, Russell IJ, Richardson GP (2012) A mouse model for human deafness DFNB22 reveals that hearing impairment is due to a loss of inner hair cell stimulation. Proc Natl Acad Sci U S A 109(47):19351–19356. doi:10.1073/pnas.1210159109
Lynch ED, Lee MK, Morrow JE, Welcsh PL, Leon PE, King MC (1997) Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science 278(5341):1315–1318
Lynn BD, Tress O, May D, Willecke K, Nagy JI (2011) Ablation of connexin30 in transgenic mice alters expression patterns of connexin26 and connexin32 in glial cells and leptomeninges. Eur J Neurosci 34(11):1783–1793. doi:10.1111/j.1460-9568.2011.07900.x
Manor U, Disanza A, Grati M, Andrade L, Lin H, Di Fiore PP, Scita G, Kachar B (2011) Regulation of stereocilia length by myosin XVa and whirlin depends on the actin-regulatory protein Eps8. Curr Biol 21(2):167–172. doi:10.1016/j.cub.2010.12.046
Mao Y, Mulvaney J, Zakaria S, Yu T, Morgan KM, Allen S, Basson MA, Francis-West P, Irvine KD (2011) Characterization of a Dchs1 mutant mouse reveals requirements for Dchs1-Fat4 signaling during mammalian development. Development 138(5):947–957. doi:10.1242/dev.057166
Marcotti W, Corns LF, Desmonds T, Kirkwood NK, Richardson GP, Kros CJ (2014) Transduction without tip links in cochlear hair cells is mediated by ion channels with permeation properties distinct from those of the mechano-electrical transducer channel. J Neurosci 34(16):5505–5514. doi:10.1523/JNEUROSCI.4086-13.2014
Marcus DC, Wu T, Wangemann P, Kofuji P (2002) KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am J Physiol Cell Physiol 282(2):C403–C407. doi:10.1152/ajpcell.00312.2001
May-Simera H, Kelley MW (2012) Planar cell polarity in the inner ear. Curr Top Dev Biol 101:111–140. doi:10.1016/B978-0-12-394592-1.00006-5
May-Simera HL, Kai M, Hernandez V, Osborn DP, Tada M, Beales PL (2010) Bbs8, together with the planar cell polarity protein Vangl2, is required to establish left-right asymmetry in zebrafish. Dev Biol 345(2):215–225. doi:10.1016/j.ydbio.2010.07.013
Mburu P, Kikkawa Y, Townsend S, Romero R, Yonekawa H, Brown SD (2006) Whirlin complexes with p55 at the stereocilia tip during hair cell development. Proc Natl Acad Sci U S A 103(29):10973–10978. doi:10.1073/pnas.0600923103
Mburu P, Mustapha M, Varela A, Weil D, El-Amraoui A, Holme RH, Rump A, Hardisty RE, Blanchard S, Coimbra RS, Perfettini I, Parkinson N, Mallon AM, Glenister P, Rogers MJ, Paige AJ, Moir L, Clay J, Rosenthal A, Liu XZ, Blanco G, Steel KP, Petit C, Brown SD (2003) Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat Genet 34(4):421–428. doi:10.1038/ng1208
Mburu P, Romero MR, Hilton H, Parker A, Townsend S, Kikkawa Y, Brown SD (2010) Gelsolin plays a role in the actin polymerization complex of hair cell stereocilia. PLoS One 5(7):e11627. doi:10.1371/journal.pone.0011627
McGee J, Goodyear RJ, McMillan DR, Stauffer EA, Holt JR, Locke KG, Birch DG, Legan PK, White PC, Walsh EJ, Richardson GP (2006) The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J Neurosci 26(24):6543–6553. doi:10.1523/JNEUROSCI.0693-06.2006
Melichar I, Syka J (1987) Electrophysiological measurements of the stria vascularis potentials in vivo. Hear Res 25(1):35–43
Merritt RC, Manor U, Salles FT, Grati M, Dose AC, Unrath WC, Quintero OA, Yengo CM, Kachar B (2012) Myosin IIIB uses an actin-binding motif in its espin-1 cargo to reach the tips of actin protrusions. Curr Biol 22(4):320–325. doi:10.1016/j.cub.2011.12.053
Merte J, Jensen D, Wright K, Sarsfield S, Wang Y, Schekman R, Ginty DD (2010) Sec24b selectively sorts Vangl2 to regulate planar cell polarity during neural tube closure. Nat Cell Biol 12(1):41–46. doi:10.1038/ncb2002, sup pp 41–48
Michalski N, Michel V, Bahloul A, Lefevre G, Barral J, Yagi H, Chardenoux S, Weil D, Martin P, Hardelin JP, Sato M, Petit C (2007) Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J Neurosci 27(24):6478–6488. doi:10.1523/JNEUROSCI.0342-07.2007
Michalski N, Michel V, Caberlotto E, Lefevre GM, van Aken AF, Tinevez JY, Bizard E, Houbron C, Weil D, Hardelin JP, Richardson GP, Kros CJ, Martin P, Petit C (2009) Harmonin-b, an actin-binding scaffold protein, is involved in the adaptation of mechanoelectrical transduction by sensory hair cells. Pflugers Arch 459(1):115–130. doi:10.1007/s00424-009-0711-x
Michel V, Goodyear RJ, Weil D, Marcotti W, Perfettini I, Wolfrum U, Kros CJ, Richardson GP, Petit C (2005) Cadherin 23 is a component of the transient lateral links in the developing hair bundles of cochlear sensory cells. Dev Biol 280(2):281–294. doi:10.1016/j.ydbio.2005.01.014
Montcouquiol M, Rachel RA, Lanford PJ, Copeland NG, Jenkins NA, Kelley MW (2003) Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature 423(6936):173–177. doi:10.1038/nature01618
Morell RJ, Friderici KH, Wei S, Elfenbein JL, Friedman TB, Fisher RA (2000) A new locus for late-onset, progressive, hereditary hearing loss DFNA20 maps to 17q25. Genomics 63(1):1–6. doi:10.1006/geno.1999.6058
Morgan CP, Barr-Gillespie PG (2013) Mechanotransduction: the elusive hair cell transduction channel revealed? Curr Biol 23(19):R887–R890. doi:10.1016/j.cub.2013.08.031
Morin M, Bryan KE, Mayo-Merino F, Goodyear R, Mencia A, Modamio-Hoybjor S, del Castillo I, Cabalka JM, Richardson G, Moreno F, Rubenstein PA, Moreno-Pelayo MA (2009) In vivo and in vitro effects of two novel gamma-actin (ACTG1) mutations that cause DFNA20/26 hearing impairment. Hum Mol Genet 18(16):3075–3089. doi:10.1093/hmg/ddp249
Nakano Y, Kim SH, Kim HM, Sanneman JD, Zhang Y, Smith RJ, Marcus DC, Wangemann P, Nessler RA, Banfi B (2009) A claudin-9-based ion permeability barrier is essential for hearing. PLoS Genet 5(8):e1000610. doi:10.1371/journal.pgen.1000610
Narimatsu M, Bose R, Pye M, Zhang L, Miller B, Ching P, Sakuma R, Luga V, Roncari L, Attisano L, Wrana JL (2009) Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell 137(2):295–307. doi:10.1016/j.cell.2009.02.025
Nayak G, Goodyear RJ, Legan PK, Noda M, Richardson GP (2011) Evidence for multiple, developmentally regulated isoforms of Ptprq on hair cells of the inner ear. Dev Neurobiol 71(2):129–141. doi:10.1002/dneu.20831
Naz S, Griffith AJ, Riazuddin S, Hampton LL, Battey JF Jr, Khan SN, Wilcox ER, Friedman TB (2004) Mutations of ESPN cause autosomal recessive deafness and vestibular dysfunction. J Med Genet 41(8):591–595. doi:10.1136/jmg.2004.018523
Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P (1997) A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange–Nielsen cardioauditory syndrome. Nat Genet 15(2):186–189. doi:10.1038/ng0297-186
Nunes FD, Lopez LN, Lin HW, Davies C, Azevedo RB, Gow A, Kachar B (2006) Distinct subdomain organization and molecular composition of a tight junction with adherens junction features. J Cell Sci 119(Pt 23):4819–4827. doi:10.1242/jcs.03233
Offner FF, Dallos P, Cheatham MA (1987) Positive endocochlear potential: mechanism of production by marginal cells of stria vascularis. Hear Res 29(2–3):117–124
Ohmori H (1985) Mechano-electrical transduction currents in isolated vestibular hair cells of the chick. J Physiol 359:189–217
Ortolano S, Di Pasquale G, Crispino G, Anselmi F, Mammano F, Chiorini JA (2008) Coordinated control of connexin 26 and connexin 30 at the regulatory and functional level in the inner ear. Proc Natl Acad Sci U S A 105(48):18776–18781. doi:10.1073/pnas.0800831105
Pallares-Ruiz N, Blanchet P, Mondain M, Claustres M, Roux AF (2002) A large deletion including most of GJB6 in recessive non syndromic deafness: a digenic effect? Eur J Hum Genet 10(1):72–76. doi:10.1038/sj.ejhg.5200762
Pan B, Geleoc GS, Asai Y, Horwitz GC, Kurima K, Ishikawa K, Kawashima Y, Griffith AJ, Holt JR (2013) TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron 79(3):504–515. doi:10.1016/j.neuron.2013.06.019
Paudyal A, Damrau C, Patterson VL, Ermakov A, Formstone C, Lalanne Z, Wells S, Lu X, Norris DP, Dean CH, Henderson DJ, Murdoch JN (2010) The novel mouse mutant, chuzhoi, has disruption of Ptk7 protein and exhibits defects in neural tube, heart and lung development and abnormal planar cell polarity in the ear. BMC Dev Biol 10:87. doi:10.1186/1471-213X-10-87
Peng AW, Belyantseva IA, Hsu PD, Friedman TB, Heller S (2009) Twinfilin 2 regulates actin filament lengths in cochlear stereocilia. J Neurosci 29(48):15083–15088. doi:10.1523/JNEUROSCI.2782-09.2009
Pepermans E, Michel V, Goodyear RJ, Bonnet C, Abdi S, Dupont T, Gherbi S, Holder M, Makrelouf M, Hardelin JP, Marlin S, Zenati A, Richardson GP, Avan P, Bahloul A, Petit C (2014) The CD2 isoform of protocadherin-15 is an essential component of the tip-link complex in mature auditory hair cells. EMBO Mol Med. doi:10.15252/emmm.201403976
Perrin BJ, Sonnemann KJ, Ervasti JM (2010) beta-actin and gamma-actin are each dispensable for auditory hair cell development but required for Stereocilia maintenance. PLoS Genet 6(10):e1001158. doi:10.1371/journal.pgen.1001158
Perrin BJ, Strandjord DM, Narayanan P, Henderson DM, Johnson KR, Ervasti JM (2013) beta-Actin and fascin-2 cooperate to maintain stereocilia length. J Neurosci 33(19):8114–8121. doi:10.1523/JNEUROSCI.0238-13.2013
Petit C (2001) Usher syndrome: from genetics to pathogenesis. Annu Rev Genomics Hum Genet 2:271–297. doi:10.1146/annurev.genom.2.1.271
Pickles JO, Comis SD, Osborne MP (1984) Cross-links between stereocilia in the guinea pig organ of Corti, and their possible relation to sensory transduction. Hear Res 15(2):103–112
Probst FJ, Fridell RA, Raphael Y, Saunders TL, Wang A, Liang Y, Morell RJ, Touchman JW, Lyons RH, Noben-Trauth K, Friedman TB, Camper SA (1998) Correction of deafness in shaker-2 mice by an unconventional myosin in a BAC transgene. Science 280(5368):1444–1447
Procaccio V, Salazar G, Ono S, Styers ML, Gearing M, Davila A, Jimenez R, Juncos J, Gutekunst CA, Meroni G, Fontanella B, Sontag E, Sontag JM, Faundez V, Wainer BH (2006) A mutation of beta -actin that alters depolymerization dynamics is associated with autosomal dominant developmental malformations, deafness, and dystonia. Am J Hum Genet 78(6):947–960. doi:10.1086/504271
Prosser HM, Rzadzinska AK, Steel KP, Bradley A (2008) Mosaic complementation demonstrates a regulatory role for myosin VIIa in actin dynamics of stereocilia. Mol Cell Biol 28(5):1702–1712. doi:10.1128/MCB.01282-07
Prost J, Barbetta C, Joanny JF (2007) Dynamical control of the shape and size of stereocilia and microvilli. Biophys J 93(4):1124–1133. doi:10.1529/biophysj.106.098038
Rehman AU, Santos-Cortez RL, Morell RJ, Drummond MC, Ito T, Lee K, Khan AA, Basra MA, Wasif N, Ayub M, Ali RA, Raza SI, Nickerson DA, Shendure J, Bamshad M, Riazuddin S, Billington N, Khan SN, Friedman PL, Griffith AJ, Ahmad W, Leal SM, Friedman TB (2014) Mutations in TBC1D24, a gene associated with epilepsy, also cause nonsyndromic deafness DFNB86. Am J Hum Genet 94(1):144–152. doi:10.1016/j.ajhg.2013.12.004
Reiners J, Marker T, Jurgens K, Reidel B, Wolfrum U (2005) Photoreceptor expression of the Usher syndrome type 1 protein protocadherin 15 (USH1F) and its interaction with the scaffold protein harmonin (USH1C). Mol Vis 11:347–355
Riazuddin S, Belyantseva IA, Giese AP, Lee K, Indzhykulian AA, Nandamuri SP, Yousaf R, Sinha GP, Lee S, Terrell D, Hegde RS, Ali RA, Anwar S, Andrade-Elizondo PB, Sirmaci A, Parise LV, Basit S, Wali A, Ayub M, Ansar M, Ahmad W, Khan SN, Akram J, Tekin M, Cook T, Buschbeck EK, Frolenkov GI, Leal SM, Friedman TB, Ahmed ZM (2012) Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet 44(11):1265–1271. doi:10.1038/ng.2426
Ricci AJ, Kennedy HJ, Crawford AC, Fettiplace R (2005) The transduction channel filter in auditory hair cells. J Neurosci 25(34):7831–7839. doi:10.1523/JNEUROSCI.1127-05.2005
Richardson GP, Russell IJ, Duance VC, Bailey AJ (1987) Polypeptide composition of the mammalian tectorial membrane. Hear Res 25(1):45–60
Rodriguez-Paris J, Schrijver I (2009) The digenic hypothesis unraveled: the GJB6 del(GJB6-D13S1830) mutation causes allele-specific loss of GJB2 expression in cis. Biochem Biophys Res Commun 389(2):354–359. doi:10.1016/j.bbrc.2009.08.152
Ross AJ, May-Simera H, Eichers ER, Kai M, Hill J, Jagger DJ, Leitch CC, Chapple JP, Munro PM, Fisher S, Tan PL, Phillips HM, Leroux MR, Henderson DJ, Murdoch JN, Copp AJ, Eliot MM, Lupski JR, Kemp DT, Dollfus H, Tada M, Katsanis N, Forge A, Beales PL (2005) Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet 37(10):1135–1140. doi:10.1038/ng1644
Russell IJ, Cody AR, Richardson GP (1986) The responses of inner and outer hair cells in the basal turn of the guinea-pig cochlea and in the mouse cochlea grown in vitro. Hear Res 22:199–216
Russell IJ, Legan PK, Lukashkina VA, Lukashkin AN, Goodyear RJ, Richardson GP (2007) Sharpened cochlear tuning in a mouse with a genetically modified tectorial membrane. Nat Neurosci 10(2):215–223. doi:10.1038/nn1828
Rzadzinska AK, Nevalainen EM, Prosser HM, Lappalainen P, Steel KP (2009) MyosinVIIa interacts with Twinfilin-2 at the tips of mechanosensory stereocilia in the inner ear. PLoS One 4(9):e7097. doi:10.1371/journal.pone.0007097
Rzadzinska AK, Schneider ME, Davies C, Riordan GP, Kachar B (2004) An actin molecular treadmill and myosins maintain stereocilia functional architecture and self-renewal. J Cell Biol 164(6):887–897. doi:10.1083/jcb.200310055
Saburi S, Hester I, Fischer E, Pontoglio M, Eremina V, Gessler M, Quaggin SE, Harrison R, Mount R, McNeill H (2008) Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat Genet 40(8):1010–1015. doi:10.1038/ng.179
Sahly I, Dufour E, Schietroma C, Michel V, Bahloul A, Perfettini I, Pepermans E, Estivalet A, Carette D, Aghaie A, Ebermann I, Lelli A, Iribarne M, Hardelin JP, Weil D, Sahel JA, El-Amraoui A, Petit C (2012) Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice. J Cell Biol 199(2):381–399. doi:10.1083/jcb.201202012
Sakaguchi H, Tokita J, Naoz M, Bowen-Pope D, Gov NS, Kachar B (2008) Dynamic compartmentalization of protein tyrosine phosphatase receptor Q at the proximal end of stereocilia: implication of myosin VI-based transport. Cell Motil Cytoskeleton 65(7):528–538. doi:10.1002/cm.20275
Salles FT, Andrade LR, Tanda S, Grati M, Plona KL, Gagnon LH, Johnson KR, Kachar B, Berryman MA (2014) CLIC5 stabilizes membrane-actin filament linkages at the base of hair cell stereocilia in a molecular complex with radixin, taperin, and myosin VI. Cytoskeleton (Hoboken) 71(1):61–78. doi:10.1002/cm.21159
Salles FT, Merritt RC Jr, Manor U, Dougherty GW, Sousa AD, Moore JE, Yengo CM, Dose AC, Kachar B (2009) Myosin IIIa boosts elongation of stereocilia by transporting espin 1 to the plus ends of actin filaments. Nat Cell Biol 11(4):443–450. doi:10.1038/ncb1851
Salt AN, Konishi T (1979) Effects of noise on cochlear potentials and endolymph potassium concentration recorded with potassium-selective electrodes. Hear Res 1(4):343–363
Salt AN, Melichar I, Thalmann R (1987) Mechanisms of endocochlear potential generation by stria vascularis. Laryngoscope 97(8 Pt 1):984–991
Schneider ME, Belyantseva IA, Azevedo RB, Kachar B (2002) Rapid renewal of auditory hair bundles. Nature 418(6900):837–838. doi:10.1038/418837a
Schneider ME, Dose AC, Salles FT, Chang W, Erickson FL, Burnside B, Kachar B (2006) A new compartment at stereocilia tips defined by spatial and temporal patterns of myosin IIIa expression. J Neurosci 26(40):10243–10252. doi:10.1523/JNEUROSCI.2812-06.2006
Schoen CJ, Burmeister M, Lesperance MM (2013) Diaphanous homolog 3 (Diap3) overexpression causes progressive hearing loss and inner hair cell defects in a transgenic mouse model of human deafness. PLoS One 8(2):e56520. doi:10.1371/journal.pone.0056520
Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, Rubie C, Hordt M, Towbin JA, Borggrefe M, Assmann G, Qu X, Somberg JC, Breithardt G, Oberti C, Funke H (1997) KCNE1 mutations cause jervell and Lange-Nielsen syndrome. Nat Genet 17(3):267–268. doi:10.1038/ng1197-267
Sekerkova G, Richter CP, Bartles JR (2011) Roles of the espin actin-bundling proteins in the morphogenesis and stabilization of hair cell stereocilia revealed in CBA/CaJ congenic jerker mice. PLoS Genet 7(3):e1002032. doi:10.1371/journal.pgen.1002032
Self T, Mahony M, Fleming J, Walsh J, Brown SD, Steel KP (1998) Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 125(4):557–566
Self T, Sobe T, Copeland NG, Jenkins NA, Avraham KB, Steel KP (1999) Role of myosin VI in the differentiation of cochlear hair cells. Dev Biol 214(2):331–341. doi:10.1006/dbio.1999.9424
Senften M, Schwander M, Kazmierczak P, Lillo C, Shin JB, Hasson T, Geleoc GS, Gillespie PG, Williams D, Holt JR, Muller U (2006) Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J Neurosci 26(7):2060–2071. doi:10.1523/JNEUROSCI.4251-05.2006
Shepherd GM, Corey DP (1994) The extent of adaptation in bullfrog saccular hair cells. J Neurosci 14(10):6217–6229
Shin JB, Krey JF, Hassan A, Metlagel Z, Tauscher AN, Pagana JM, Sherman NE, Jeffery ED, Spinelli KJ, Zhao H, Wilmarth PA, Choi D, David LL, Auer M, Barr-Gillespie PG (2013) Molecular architecture of the chick vestibular hair bundle. Nat Neurosci 16(3):365–374. doi:10.1038/nn.3312
Shin JB, Streijger F, Beynon A, Peters T, Gadzala L, McMillen D, Bystrom C, Van der Zee CE, Wallimann T, Gillespie PG (2007) Hair bundles are specialized for ATP delivery via creatine kinase. Neuron 53(3):371–386. doi:10.1016/j.neuron.2006.12.021
Siemens J, Lillo C, Dumont RA, Reynolds A, Williams DS, Gillespie PG, Muller U (2004) Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature 428(6986):950–955. doi:10.1038/nature02483
Simmler MC, Zwaenepoel II, Verpy E, Guillaud L, Elbaz C, Petit C, Panthier JJ (2000) Twister mutant mice are defective for otogelin, a component specific to inner ear acellular membranes. Mamm Genome 11(11):961–966. doi:10.1007/s003350010197
Sipe CW, Lu X (2011) Kif3a regulates planar polarization of auditory hair cells through both ciliary and non-ciliary mechanisms. Development 138(16):3441–3449. doi:10.1242/dev.065961
Sollner C, Rauch GJ, Siemens J, Geisler R, Schuster SC, Muller U, Nicolson T (2004) Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature 428(6986):955–959. doi:10.1038/nature02484
Spiden SL, Bortolozzi M, Di Leva F, de Angelis MH, Fuchs H, Lim D, Ortolano S, Ingham NJ, Brini M, Carafoli E, Mammano F, Steel KP (2008) The novel mouse mutation Oblivion inactivates the PMCA2 pump and causes progressive hearing loss. PLoS Genet 4(10):e1000238. doi:10.1371/journal.pgen.1000238
Stapelbroek JM, Peters TA, van Beurden DH, Curfs JH, Joosten A, Beynon AJ, van Leeuwen BM, van der Velden LM, Bull L, Oude Elferink RP, van Zanten BA, Klomp LW, Houwen RH (2009) ATP8B1 is essential for maintaining normal hearing. Proc Natl Acad Sci U S A 106(24):9709–9714. doi:10.1073/pnas.0807919106
Stauffer EA, Scarborough JD, Hirono M, Miller ED, Shah K, Mercer JA, Holt JR, Gillespie PG (2005) Fast adaptation in vestibular hair cells requires myosin-1c activity. Neuron 47(4):541–553. doi:10.1016/j.neuron.2005.07.024
Stepanyan R, Frolenkov GI (2009) Fast adaptation and Ca2+ sensitivity of the mechanotransducer require myosin-XVa in inner but not outer cochlear hair cells. J Neurosci 29(13):4023–4034. doi:10.1523/JNEUROSCI.4566-08.2009
Tarchini B, Jolicoeur C, Cayouette M (2013) A molecular blueprint at the apical surface establishes planar asymmetry in cochlear hair cells. Dev Cell 27(1):88–102. doi:10.1016/j.devcel.2013.09.011
Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Sohl G, Jahnke K, Winterhager E, Herberhold C, Hardelin JP, Petit C, Willecke K (2003) Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet 12(1):13–21
Thalmann I, Thallinger G, Crouch EC, Comegys TH, Barrett N, Thalmann R (1987) Composition and supramolecular organization of the tectorial membrane. Laryngoscope 97(3 Pt 1):357–367
Tilney LG, Derosier DJ, Mulroy MJ (1980) The organization of actin filaments in the stereocilia of cochlear hair cells. J Cell Biol 86(1):244–259
Tilney MS, Tilney LG, Stephens RE, Merte C, Drenckhahn D, Cotanche DA, Bretscher A (1989) Preliminary biochemical characterization of the stereocilia and cuticular plate of hair cells of the chick cochlea. J Cell Biol 109(4 Pt 1):1711–1723
Torban E, Patenaude AM, Leclerc S, Rakowiecki S, Gauthier S, Andelfinger G, Epstein DJ, Gros P (2008) Genetic interaction between members of the Vangl family causes neural tube defects in mice. Proc Natl Acad Sci U S A 105(9):3449–3454. doi:10.1073/pnas.0712126105
Tyson J, Tranebjaerg L, Bellman S, Wren C, Taylor JF, Bathen J, Aslaksen B, Sorland SJ, Lund O, Malcolm S, Pembrey M, Bhattacharya S, Bitner-Glindzicz M (1997) IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum Mol Genet 6(12):2179–2185
van Wijk E, Krieger E, Kemperman MH, De Leenheer EM, Huygen PL, Cremers CW, Cremers FP, Kremer H (2003) A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J Med Genet 40(12):879–884
van Wijk E, van der Zwaag B, Peters T, Zimmermann U, Te Brinke H, Kersten FF, Marker T, Aller E, Hoefsloot LH, Cremers CW, Cremers FP, Wolfrum U, Knipper M, Roepman R, Kremer H (2006) The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum Mol Genet 15(5):751–765. doi:10.1093/hmg/ddi490
Verpy E, Leibovici M, Michalski N, Goodyear RJ, Houdon C, Weil D, Richardson GP, Petit C (2011) Stereocilin connects outer hair cell stereocilia to one another and to the tectorial membrane. J Comp Neurol 519(2):194–210. doi:10.1002/cne.22509
Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJ, Slim R, Petit C (2000) A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet 26(1):51–55. doi:10.1038/79171
Verpy E, Masmoudi S, Zwaenepoel I, Leibovici M, Hutchin TP, Del Castillo I, Nouaille S, Blanchard S, Laine S, Popot JL, Moreno F, Mueller RF, Petit C (2001) Mutations in a new gene encoding a protein of the hair bundle cause non-syndromic deafness at the DFNB16 locus. Nat Genet 29(3):345–349. doi:10.1038/ng726
Verpy E, Weil D, Leibovici M, Goodyear RJ, Hamard G, Houdon C, Lefevre GM, Hardelin JP, Richardson GP, Avan P, Petit C (2008) Stereocilin-deficient mice reveal the origin of cochlear waveform distortions. Nature 456(7219):255–258. doi:10.1038/nature07380
Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J (1996) Inner ear defects induced by null mutation of the isk gene. Neuron 17(6):1251–1264
Walsh T, Pierce SB, Lenz DR, Brownstein Z, Dagan-Rosenfeld O, Shahin H, Roeb W, McCarthy S, Nord AS, Gordon CR, Ben-Neriah Z, Sebat J, Kanaan M, Lee MK, Frydman M, King MC, Avraham KB (2010) Genomic duplication and overexpression of TJP2/ZO-2 leads to altered expression of apoptosis genes in progressive nonsyndromic hearing loss DFNA51. Am J Hum Genet 87(1):101–109. doi:10.1016/j.ajhg.2010.05.011
Walsh T, Shahin H, Elkan-Miller T, Lee MK, Thornton AM, Roeb W, Abu Rayyan A, Loulus S, Avraham KB, King MC, Kanaan M (2010) Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am J Hum Genet 87(1):90–94. doi:10.1016/j.ajhg.2010.05.010
Walsh T, Walsh V, Vreugde S, Hertzano R, Shahin H, Haika S, Lee MK, Kanaan M, King MC, Avraham KB (2002) From flies’ eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc Natl Acad Sci U S A 99(11):7518–7523. doi:10.1073/pnas.102091699
Walsh VL, Raviv D, Dror AA, Shahin H, Walsh T, Kanaan MN, Avraham KB, King MC (2011) A mouse model for human hearing loss DFNB30 due to loss of function of myosin IIIA. Mamm Genome 22(3–4):170–177. doi:10.1007/s00335-010-9310-6
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153(4):910–918. doi:10.1016/j.cell.2013.04.025
Wang J, Mark S, Zhang X, Qian D, Yoo SJ, Radde-Gallwitz K, Zhang Y, Lin X, Collazo A, Wynshaw-Boris A, Chen P (2005) Regulation of polarized extension and planar cell polarity in the cochlea by the vertebrate PCP pathway. Nat Genet 37(9):980–985. doi:10.1038/ng1622
Wang Y, Guo N, Nathans J (2006) The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J Neurosci 26(8):2147–2156. doi:10.1523/JNEUROSCI.4698-05.2005
Warchol ME, Montcouquiol M (2010) Maintained expression of the planar cell polarity molecule Vangl2 and reformation of hair cell orientation in the regenerating inner ear. J Assoc Res Otolaryngol 11(3):395–406. doi:10.1007/s10162-010-0209-4
Watson GM, Hessinger DA (1987) Receptor-mediated endocytosis of a chemoreceptor involved in triggering the discharge of cnidae in a sea anemone tentacle. Tissue Cell 19(6):747–755
Watson GM, Mire P (2001) Reorganization of actin during repair of hair bundle mechanoreceptors. J Neurocytol 30(11):895–906
Webb SW, Grillet N, Andrade LR, Xiong W, Swarthout L, Della Santina CC, Kachar B, Muller U (2011) Regulation of PCDH15 function in mechanosensory hair cells by alternative splicing of the cytoplasmic domain. Development 138(8):1607–1617. doi:10.1242/dev.060061
Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD et al (1995) Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 374(6517):60–61. doi:10.1038/374060a0
Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, Delmaghani S, Adato A, Nadifi S, Zina ZB, Hamel C, Gal A, Ayadi H, Yonekawa H, Petit C (2003) Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet 12(5):463–471
Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ (2004) Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet 74(2):357–366. doi:10.1086/381685
Wilson SM, Householder DB, Coppola V, Tessarollo L, Fritzsch B, Lee EC, Goss D, Carlson GA, Copeland NG, Jenkins NA (2001) Mutations in Cdh23 cause nonsyndromic hearing loss in waltzer mice. Genomics 74(2):228–233. doi:10.1006/geno.2001.6554
Wu L, Pan L, Wei Z, Zhang M (2011) Structure of MyTH4-FERM domains in myosin VIIa tail bound to cargo. Science 331(6018):757–760. doi:10.1126/science.1198848
Xiong W, Grillet N, Elledge HM, Wagner TF, Zhao B, Johnson KR, Kazmierczak P, Muller U (2012) TMHS is an integral component of the mechanotransduction machinery of cochlear hair cells. Cell 151(6):1283–1295. doi:10.1016/j.cell.2012.10.041
Xu Z, Peng AW, Oshima K, Heller S (2008) MAGI-1, a candidate stereociliary scaffolding protein, associates with the tip-link component cadherin 23. J Neurosci 28(44):11269–11276. doi:10.1523/JNEUROSCI.3833-08.2008
Yagi H, Tokano H, Maeda M, Takabayashi T, Nagano T, Kiyama H, Fujieda S, Kitamura K, Sato M (2007) Vlgr1 is required for proper stereocilia maturation of cochlear hair cells. Genes Cells 12(2):235–250. doi:10.1111/j.1365-2443.2007.01046.x
Yamamoto S, Nishimura O, Misaki K, Nishita M, Minami Y, Yonemura S, Tarui H, Sasaki H (2008) Cthrc1 selectively activates the planar cell polarity pathway of Wnt signaling by stabilizing the Wnt-receptor complex. Dev Cell 15(1):23–36. doi:10.1016/j.devcel.2008.05.007
Yan J, Pan L, Chen X, Wu L, Zhang M (2010) The structure of the harmonin/sans complex reveals an unexpected interaction mode of the two Usher syndrome proteins. Proc Natl Acad Sci U S A 107(9):4040–4045. doi:10.1073/pnas.0911385107
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R (2013) One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154(6):1370–1379. doi:10.1016/j.cell.2013.08.022
Yang T, Gurrola JG 2nd, Wu H, Chiu SM, Wangemann P, Snyder PM, Smith RJ (2009) Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet 84(5):651–657. doi:10.1016/j.ajhg.2009.04.014
Yariz KO, Duman D, Seco CZ, Dallman J, Huang M, Peters TA, Sirmaci A, Lu N, Schraders M, Skromne I, Oostrik J, Diaz-Horta O, Young JI, Tokgoz-Yilmaz S, Konukseven O, Shahin H, Hetterschijt L, Kanaan M, Oonk AM, Edwards YJ, Li H, Atalay S, Blanton S, Desmidt AA, Liu XZ, Pennings RJ, Lu Z, Chen ZY, Kremer H, Tekin M (2012) Mutations in OTOGL, encoding the inner ear protein otogelin-like, cause moderate sensorineural hearing loss. Am J Hum Genet 91(5):872–882. doi:10.1016/j.ajhg.2012.09.011
Zampini V, Ruttiger L, Johnson SL, Franz C, Furness DN, Waldhaus J, Xiong H, Hackney CM, Holley MC, Offenhauser N, Di Fiore PP, Knipper M, Masetto S, Marcotti W (2011) Eps8 regulates hair bundle length and functional maturation of mammalian auditory hair cells. PLoS Biol 9(4):e1001048. doi:10.1371/journal.pbio.1001048
Zhang DS, Piazza V, Perrin BJ, Rzadzinska AK, Poczatek JC, Wang M, Prosser HM, Ervasti JM, Corey DP, Lechene CP (2012) Multi-isotope imaging mass spectrometry reveals slow protein turnover in hair–cell stereocilia. Nature 481(7382):520–524. doi:10.1038/nature10745
Zhang L, Hu L, Chai Y, Pang X, Yang T, Wu H (2014) A dominant mutation in the stereocilia-expressing gene TBC1D24 is a probable cause for nonsyndromic hearing impairment. Hum Mutat. doi:10.1002/humu.22558
Zheng J, Miller KK, Yang T, Hildebrand MS, Shearer AE, DeLuca AP, Scheetz TE, Drummond J, Scherer SE, Legan PK, Goodyear RJ, Richardson GP, Cheatham MA, Smith RJ, Dallos P (2011) Carcinoembryonic antigen-related cell adhesion molecule 16 interacts with alpha-tectorin and is mutated in autosomal dominant hearing loss (DFNA4). Proc Natl Acad Sci U S A 108(10):4218–4223. doi:10.1073/pnas.1005842108
Zheng L, Sekerkova G, Vranich K, Tilney LG, Mugnaini E, Bartles JR (2000) The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell 102(3):377–385
Zhu M, Yang T, Wei S, DeWan AT, Morell RJ, Elfenbein JL, Fisher RA, Leal SM, Smith RJ, Friderici KH (2003) Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am J Hum Genet 73(5):1082–1091. doi:10.1086/379286
Zou J, Zheng T, Ren C, Askew C, Liu XP, Pan B, Holt JR, Wang Y, Yang J (2014) Deletion of PDZD7 disrupts the Usher syndrome type 2 protein complex in cochlear hair cells and causes hearing loss in mice. Hum Mol Genet. doi:10.1093/hmg/ddt629
Acknowledgments
The authors’ research is supported by grants from the European Research council (ERC-2011-AdG 294570 HAIRBUNDLE), the Agence nationale pour la recherche (EARMEC—ANR 11 BSV5 011 02), the LABEX LIFESENSES (reference ANR-10-LABX-65) supported by French state funds within the Investissements d’Avenir programme under reference ANR-11-IDEX-0004-02, BNP Paribas and LHW Stiftung. The authors are particularly indebted to Jean-Pierre Hardelin for his help in improving the manuscript, Vincent Michel and Gaelle M. Lefèvre for the scanning electron micrographs, Crystel Bonnet for her help in completing the gene list table and Montserrat Bosch Grau, Jacques Boutet de Monvel, Vincent Michel, Amel Bahloul, Elise Pepermans, Andrea Lelli and Ménélik Labbé for their critical reading of the manuscript
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Michalski, N., Petit, C. Genetics of auditory mechano-electrical transduction. Pflugers Arch - Eur J Physiol 467, 49–72 (2015). https://doi.org/10.1007/s00424-014-1552-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-014-1552-9