
Abstract
Purpose
The aim of this study was to determine the molecular genetic basis of an early-onset severe retinal dystrophy in three unrelated consecutive patients of Czech origin and to describe their ocular phenotype.
Methods
DNA samples from two probands were analyzed using a genotyping microarray (Asper) followed by either target analysis of 43 genes implicated in retinal disorders by next generation sequencing or whole-exome sequencing, respectively. The third proband underwent conventional Sanger sequencing of CRB1 based on her ocular findings.
Results
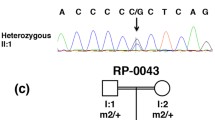
All three probands harboured a known disease-causing mutation c.2843G>A; p.(Cys948Tyr) in the CRB1 gene. One individual was homozygous for this mutation, while in the other two probands c.2308G>A; p.(Gly770Ser) and c.3121A>G; p.(Met1041Val) were also identified in the heterozygous state, respectively. Both variants were novel and evaluated by in silico analysis as pathogenic. A false-negative result was observed in one of the two samples examined by the genotyping microarray. Disease onset in all patients was before the age of 7 years. Hypermetropic refractive error, bilateral nummular retinal pigmentation, retinal thickening and cystoid spaces in the macula were observed in two probands, aged 6 and 7 years. The third proband, aged 28 years, had bone spicule-like pigmentary changes associated with increased retinal nerve fiber layer.
Conclusions
The first study reporting on the molecular genetic cause of non-syndromic early-onset severe retinal dystrophy in Czech patients identified one homozygous and two compound heterozygote probands with CRB1 mutations. Retina nerve fibre layer measurements should be considered an integral part of the clinical evaluation of retinal dystrophies. Detailed clinical examination and imaging can both direct molecular screening and help to confirm or refute disease causation of identified variants.


Similar content being viewed by others


References
Bujakowska K, Audo I, Mohand-Said S, Lancelot ME, Antonio A, Germain A, Leveillard T, Letexier M, Saraiva JP, Lonjou C, Carpentier W, Sahel JA, Bhattacharya SS, Zeitz C (2012) CRB1 mutations in inherited retinal dystrophies. Hum Mutat 33:306–315. doi:10.1002/humu.21653
den Hollander AI, Roepman R, Koenekoop RK, Cremers FP (2008) Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res 27:391–419. doi:10.1016/j.preteyeres.2008.05.003
Hartong DT, Berson EL, Dryja TP (2006) Retinitis pigmentosa. Lancet 368:1795–1809. doi:10.1016/S0140-6736(06)69740-7
Weleber RG, Michaelides M, Trzupek KM, Stover NB, Stone EM (2011) The phenotype of Severe Early Childhood Onset Retinal Dystrophy (SECORD) from mutation of RPE65 and differentiation from Leber congenital amaurosis. Invest Ophthalmol Vis Sci 52:292–302. doi:10.1167/iovs.10-6106
Daiger SP, Sullivan LS, Bowne SJ (2013) Genes and mutations causing retinitis pigmentosa. Clin Genet 84:132–141. doi:10.1111/cge.12203
Chacon-Camacho OF, Zenteno JC (2015) Review and update on the molecular basis of Leber congenital amaurosis. World J Clin Cases 3:112–124. doi:10.12998/wjcc.v3.i2.112
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. doi:10.1038/nmeth0410-248
Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, Mooney SD, Radivojac P (2009) Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25:2744–2750. doi:10.1093/bioinformatics/btp528
Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R (2009) Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat 30:1237–1244. doi:10.1002/humu.21047
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the functional effect of amino acid substitutions and indels. PLoS One 7:e46688. doi:10.1371/journal.pone.0046688
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–81. doi:10.1038/nprot.2009.86
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11:361–2. doi:10.1038/nmeth.2890
den Dunnen JT, Antonarakis SE (2001) Nomenclature for the description of human sequence variations. Hum Genet 109:121–124
den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T, Antonarakis SE, Taschner PE, Human Genome Variation Society (HGVS), the Human Variome Project (HVP) and the Human Genome Organisation (HUGO) (2016) HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. doi:10.1002/humu.22981
Notredame C, Higgins DG, Heringa J (2000) T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–17
Tosi J, Tsui I, Lima L, Wang NK, Tsang SH (2009) Case report: autofluorescence imaging and phenotypic variance in a sibling pair with early onset retinal dystrophy due to defective CRB1 function. Curr Eye Res 34:395–400. doi:10.1080/02713680902859639
Andreoli MT, Lim JI (2014) Optical coherence tomography retinal thickness and volume measurements in X-linked retinoschisis. Am J Ophthalmol 158:567–573.e2. doi:10.1016/j.ajo.2014.05.028
Kousal B, Zahlava J, Vejvalkova S, Hejtmankova M, Liskova P (2014) The molecular genetic and clinical findings in two probands with Stargardt disease. Cesk Slov Oftalmol 70:228–233
Burke TR, Tsang SH (2011) Allelic and phenotypic heterogeneity in ABCA4 mutations. Ophthalmic Genet 32:165–174. doi:10.3109/13816810.2011.565397
Sohrab MA, Allikmets R, Guarnaccia MM, Smith RT (2010) Preimplantation genetic diagnosis for stargardt disease. Am J Ophthalmol 149:651–655.e2. doi:10.1016/j.ajo.2009.11.029
Henderson RH, Waseem N, Searle R, van der Spuy J, Russell-Eggitt I, Bhattacharya SS, Thompson DA, Holder GE, Cheetham ME, Webster AR, Moore AT (2007) An assessment of the apex microarray technology in genotyping patients with Leber congenital amaurosis and early-onset severe retinal dystrophy. Invest Ophthalmol Vis Sci 48:5684–5689. doi:10.1167/iovs.07-0207
Cordovez JA, Traboulsi EI, Capasso JE, Sadagopan KA, Ganesh A, Rychwalski PJ, Neely KA, Brodie SE, Levin AV (2014) Retinal Dystrophy with Intraretinal Cystoid Spaces Associated with Mutations in the Crumbs Homologue (CRB1) gene. Ophthalmic Genet 36:257–264. doi:10.3109/13816810.2014.881505
Henderson RH, Mackay DS, Li Z, Moradi P, Sergouniotis P, Russell-Eggitt I, Thompson DA, Robson AG, Holder GE, Webster AR, Moore AT (2011) Phenotypic variability in patients with retinal dystrophies due to mutations in CRB1. Br J Ophthalmol 95:811–817. doi:10.1136/bjo.2010.186882
Jacobson SG, Cideciyan AV, Aleman TS, Pianta MJ, Sumaroka A, Schwartz SB, Smilko EE, Milam AH, Sheffield VC, Stone EM (2003) Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum Mol Genet 12:1073–1078
Acknowledgment
The authors thank Kinga Bujakowska for providing CRB1 primers.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by UNCE 204011 and PRVOUK-P24/LF1/3 programs of the Charles University in Prague and by Leontinka foundation. The sponsor had no role in the design or conduct of this research. BK is supported by SVV UK 260256/2016. MM is supported by grants from the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, the Foundation Fighting Blindness, the Special Trustees of Moorfields Eye Hospital and by an FFB Career Development Award. We thank The National Center for Medical Genomics (LM2015091) for bioinformatical support with next-generation sequencing data analysis and for providing ethnically matched population frequency data.
Conflict of interest
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers' bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 297 kb)
Rights and permissions
About this article

Cite this article
Kousal, B., Dudakova, L., Gaillyova, R. et al. Phenotypic features of CRB1-associated early-onset severe retinal dystrophy and the different molecular approaches to identifying the disease-causing variants. Graefes Arch Clin Exp Ophthalmol 254, 1833–1839 (2016). https://doi.org/10.1007/s00417-016-3358-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-016-3358-2