
Abstract
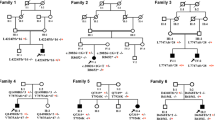
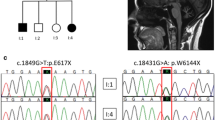
Autosomal recessive cerebellar ataxia type 3 (ARCA3) is a rare inherited disorder caused by mutations in the ANO10 gene. The disease is characterized by slowly progressive spastic ataxia variably associated with motor neuron involvement, epilepsy, and cognitive decline. We performed mutational screening in 80 patients with sporadic or autosomal recessive adult-onset ataxia. We identified 11 ANO10 gene variants in 10 patients from 8 families (10%): 4 mutations were previously described and 7 were novel. Age at onset ranged between 27 and 53 years. All patients presented ataxia, pyramidal signs and cerebellar atrophy at brain MRI. Additional signs were bradykinesia (7/10), mild vertical gaze paresis (5/10), pes cavus (4/10), and sphincteric disturbances (3/10). Six patients, with normal MMSE score, failed several neuropsychological tests rating executive functions. Three patients had giant somatosensory evoked potentials and epileptic spikes in EEG without clinical evidence of seizures. Our observational study indicates a high frequency of ARCA3 disease in sporadic patients with adult-onset cerebellar ataxia. We extended the ANO10 mutational spectrum with the identification of novel gene variants, and further defined the clinical, cognitive, and neurophysiological features in a new cohort of patients. These findings may contribute to the refinement of the complex ARCA3 phenotype and be valuable in clinical management and natural history studies.



Similar content being viewed by others

References
Parodi L, Coarelli G, Stevanin G, Brice A, Durr A (2018) Hereditary ataxias and paraparesias: clinical and genetic update. Curr Opin Neurol 31:462–471
Beaudin M, Klein CJ, Rouleau GA, Dupré N (2017) Systematic review of autosomal recessive ataxias and proposal for a classification. Cerebellum Ataxias 4:3
Vermeer S, Hoischen A, Meijer RP et al (2010) Targeted next-generation sequencing of a 12.5 Mb homozygous region reveals ANO10 mutations in patients with autosomal-recessive cerebellar ataxia. Am J Hum Genet 87:813–819
Renaud M, Anheim M, Kamsteeg EJ et al (2014) Autosomal recessive cerebellar ataxia type 3 due to ANO10 mutations: delineation and genotype-phenotype correlation study. JAMA Neurol 71:1305–1310
Balreira A, Boczonadi V, Barca E et al (2014) ANO10 mutations cause ataxia and coenzyme Q10 deficiency. J Neurol 261:2192–2198
Fogel BL, Lee H, Deignan JL et al (2014) Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol 71:1237–1246
Chamova T, Florez L, Guergueltcheva V et al (2012) ANO10 c.1150_1151del is a founder mutation causing autosomal recessive cerebellar ataxia in Roma/Gypsies. J Neurol 259:906–911
Maruyama H, Morino H, Miyamoto R, Murakami N, Hamano T, Kawakami H (2014) Exome sequencing reveals a novel ANO10 mutation in a Japanese patient with autosomal recessive spinocerebellar ataxia. Clin Genet 85:296–297
Yoshida K, Miyatake S, Kinoshita T et al (2014) ‘Cortical cerebellar atrophy’ dwindles away in the era of next-generation sequencing. J Hum Genet 59:589–590
Minnerop M, Bauer P (2015) Autosomal recessive cerebellar ataxia 3 due to homozygote c.132dupA mutation within the ANO10 gene. JAMA Neurol 72:238–239
Chamard L, Sylvestre G, Koenig M, Magnin E (2016) Executive and attentional disorders, epilepsy and porencephalic cyst in autosomal recessive cerebellar ataxia type 3 due to ANO10 mutation. Eur Neurol 75:186–190
Mišković ND, Domingo A, Dobričić V et al (2016) Seemingly dominant inheritance of a recessive ANO10 mutation in romani families with cerebellar ataxia. Mov Disord 31:1929–1931
Bodranghien F, Oulad Ben Taib N, Van Maldergem L, Manto M (2017) A postural tremor highly responsive to transcranial cerebello-cerebral DCS in ARCA3. Front Neurol 8:71
Sun M, Johnson AK, Nelakuditi V et al (2018) Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet Med. https://doi.org/10.1038/s41436-018-0007-7
Coutelier M, Hammer MB, Stevanin G et al (2018) Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol 75:591–599
Richards S, Aziz N, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Halliday DM, Rosenberg JR, Amjad AM, Breeze P, Conway BA, Farmer SF (1995) A framework for the analysis of mixed time series/point process data—theory and application to the study of physiological tremor, single motor unit discharges and electromyograms. Prog Biophys Mol Biol 64:237–278
Panzica F, Canafoglia L, Franceschetti S et al (2003) Movement-activated myoclonus in genetically defined progressive myoclonic epilepsies: EEG-EMG relationship estimated using autoregressive models. Clin Neurophysiol 114:1041–1052
Mochizuki H, Hanajima R, Kowa H et al (2001) Somatosensory evoked potential recovery in myotonic dystrophy. Clin Neurophysiol 112:793–799
Visani E, Canafoglia L, Rossi Sebastiano D et al (2013) Giant SEPs and SEP-recovery function in Unverricht–Lundborg disease. Clin Neurophysiol 124:1013–1018
Synofzik M, Smets K, Mallaret M et al (2016) SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: a large multi-centre study. Brain 139:1378–1393
Slapik M, Kronemer SI, Morgan O et al (2018) Visuospatial organization and recall in cerebellar ataxia. Cerebellum. https://doi.org/10.1007/s12311-018-0948-z
Schmahmann JD, Sherman JC (1998) The cerebellar cognitive affective syndrome. Brain 121:561–579
Saccà F, Costabile T, Abate F et al (2017) Normalization of timed neuropsychological tests with the PATA rate and nine-hole pegboard tests. J Neuropsychol 12:471–483
Martín-Palomeque G, Castro-Ortiz A, Pamplona-Valenzuela P, Saiz-Sepúlveda M, Cabañes-Martínez L, López JR (2017) Large amplitude cortical evoked potentials in nonepileptic patients. Reviving an old neurophysiologic tool to help detect CNS pathology. J Clin Neurophysiol 34:84–91
Miwa H, Mizuno Y (2002) Enlargements of somatosensory-evoked potentials in progressive supranuclear palsy. Acta Neurol Scand 106:209–212
Shimizu T, Bokuda K, Kimura H et al (2018) Sensory cortex hyperexcitability predicts short survival in amyotrophic lateral sclerosis. Neurology 90:1578–1587
Acknowledgements
This work was supported in part by research grants from the Italian Ministry of Health: RF-2011-02351165 (to F.T.), RF-2011-02347420 (to C.M.)
Funding
Research Grant RF-2011-02351165 from the Italian Ministry of Health to F.T., and RF-2011-02347420 to C.M.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
All authors declare that they have no conflict of interest.
Ethical standard
All patients gave written informed consent for the clinical and genetic tests in agreement with the procedures approved by the Local Ethic Committee. The consent forms routinely used in our Hospital specifically enquire the patient consent for diagnostic and research purposes. Ethics committee approval is not required for retrospective anonymized observational studies.
Rights and permissions
About this article

Cite this article
Nanetti, L., Sarto, E., Castaldo, A. et al. ANO10 mutational screening in recessive ataxia: genetic findings and refinement of the clinical phenotype. J Neurol 266, 378–385 (2019). https://doi.org/10.1007/s00415-018-9141-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-9141-z