
Abstract
Background
From the start of the pandemic, many European otolaryngologists observed an unprecendented number of anosmic patients. Early reports proposed that anosmia could be the first or even the only symptom of COVID-19 infection, prompting calls for self-isolation in affected patients.
Methods
In the present article, we review the COVID-19 anosmia literature and try to answer the following two questions: first, why is COVID-19 infection responsible for such a high incidence of anosmia? Second, in patients with more severe forms is anosmia really less prevalent and why?
Results
In terms of the etiology of olfactory dysfunction, several hypotheses were proposed at the outset of the pandemic; that olfactory cleft inflammation and obstruction caused a localized conductive loss, that there was injury to the sustentacular supporting cells in the olfactory epithelium or, given the known neurotropic potential of coronavirus, that the virus could invade and damage the olfactory bulb. Olfactory cleft obstruction may contribute to the olfactory dysfunction in some patients, perhaps most likely in those that show very early resolution, it cannot account for the loss in all patients.
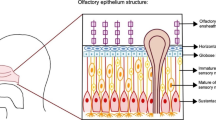
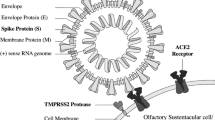
Moreover, disordered regrowth and a predominance of immature neurons have been shown to be associated with parosmia, which is a common finding amongst patients with Covid-related anosmia. A central mechanism therefore certainly seems to be consistent with the group of patients with more prolonged olfactory deficits. Sustentacular cells showing ACE-2 immunohistochemical expression 200 to 700 times greater than nasal or tracheal epithelia seem to be the main SARS-CoV-2 gateway.
As the pathophysiology of COVID-19 anosmia seems to be better understood, the question of why patients with a moderate to severe form of COVID-19 infection have less olfactory involvement remains unresolved. Different potential explanations are discussed in this review.
Conclusions
The last 5 months have benefited from great international collaborative research, first highlighting and then proving the value of loss of smell and taste as a symptom of COVID-19. Adoption of loss of smell into the case definition by international public health bodies will facilitate control of disease transmission.
Similar content being viewed by others
Dear Editor,
From the start of the pandemic, many European otolaryngologists observed an unprecedented number of anosmic patients. Early reports proposed that anosmia could be the first or even the only symptom of COVID-19 infection, prompting calls for self-isolation in affected patients [1]. In this context, we carried out the first European study describing the importance of this symptom among patients with a mild form of COVID-19 infection, where we described that 86% of 417 patients with a mild to moderate form of COVID-19 (proven by polymerase chain reaction (PCR) testing) presented with anosmia and 88% dysgeusia [2]. In a subsequent series of 2013 patients with a mild to moderate form of COVID-19, the importance of this symptom was confirmed, since anosmia was the most frequent symptom affecting 87% of patients [3].
We then looked to address the next important question—what proportion of patients presenting with isolated and/or initial severe anosmia, with or without additional upper respiratory symptoms during the COVID pandemic, were actually COVID-positive? To answer this question, we conducted a prospective study enlisting patients with recent and severe anosmia in the weeks following our first observations. First, we carried out a study of 78 patients with sudden and initial anosmia. We were thus able to show that if the swab was performed within the first 14 days of symptoms, 87% of these anosmic patients were COVID-19 + [4]. Using serological testing (Zentec kit), we confirmed with the group showing symptoms for more than 14 days that 93% of anosmic patients were COVID-19 + .
Our study then turned to psychophysical measurements of olfaction using ‘Sniffin Sticks’, which were carried out in 86 patients with mild forms of COVID-19 associated with subjective anosmia [5] and 47 more severely affected patients who were hospitalized. In mild to moderate COVID-19 patients managed as outpatients, this identified 41 anosmic (47.7%), 12 hyposmic (14.0%), and 33 normosmic (38.3%) patients.[5]. In contrast, the psychophysical olfactory evaluations of hospitalized patients reported that 4 (8.5%) and 9 (19.1%) patients reported anosmia and hyposmia. However, testing in hospitalized patients was, by necessity, performed at a later time point in the disease process, potentially allowing for recovery. Romero-Sanchez and colleagues described on a large clinical series of 841 patients hospitalized with COVID-19 that 57.4% developed some form of neurological symptom. Among these, anosmia concerned only 5% of patients [6].
While self-reported new onset loss of smell is important from an infection control perspective, as it allows patients to recognize COVID infection and self-isolate at an early stage; however, psychophysical testing has demonstrated that self-reporting underestimates the true prevalence of olfactory dysfunction [7, 8]. Most self-reported questionnaires combine smell and taste disorders, due to the difficulty in distinguishing perception of true taste and flavor, Psychophysical gustatory tests confirm the presence of true gustatory dysfunction in nearly half of patients tested, but with mean scores returning to normal within 3 weeks of onset [9]. Finally, preliminary data concerning the recovery from anosmia also provide some useful observations in understanding the physiopathology of this condition.
First, we studied the self-reported early recovery of anosmia in a clinical series of 382 patients. 86.4% reported complete anosmia and a further 11.5% a very severe loss of smell at the time of completing the first survey. At follow-up 1 week later, 80.1% reported lower severity scores at follow-up, 17.6% were unchanged and 1.9% had deteriorated [10]. Subsequently, in a prospective clinical series of 88 COVID-19 self-reported anosmic patients, we observed that 79.5% of patients recovered in the 2 first months following onset. According to the psychophysical olfactory evaluations; 25.0% rapidly recovered normal olfactory function within the first 15 days following the onset of anosmia, while 10.2% required 16–30 days, 28.4% recovered between day 31—45, and 15.9% between 46–70 days after onset. We demonstrated that baseline severity of olfactory loss was predictive for persistent loss. We have also shown that self-reporting overestimates true recovery rates, with patients self-reporting complete recovery being found to have persistent mild to moderate hyposmia on psychophysical testing [11].
These various observations led us to ask two further important questions: first, why is COVID-19 infection responsible for such a high incidence of anosmia? Second, in patients with more severe forms is anosmia really less prevalent?
In terms of the etiology of olfactory dysfunction, several hypotheses were proposed at the outset of the pandemic; that olfactory cleft inflammation and obstruction caused a localized conductive loss, that there was injury to the sustentacular supporting cells in the olfactory epithelium or, given the known neurotropic potential of coronavirus, that the virus could invade and damage the olfactory bulb. To address at the first question, we performed sinus computed tomography (CT) on 16 patients with persistent anosmia on psychophysical olfactory evaluation. Surprisingly, we found that the olfactory cleft was completely clear in 7 patients, while only 3 patients were completely opacified on CT imaging, and partially opacified in the remaining 6 (Fig. 1). The sinuses and nasal cavities showed no evidence of mucosal thickening. This suggests that while olfactory cleft obstruction may contribute to the olfactory dysfunction in some patients, perhaps most likely in those that show very early resolution, it cannot account for the loss in all patients. Chen and colleagues have studied the immunohistochemical expression of ACE-2, the SARS-CoV 2 target receptor protein, in 11 specimens of neuro-olfactory epithelia [12]. They showed that ACE-2 immunohistochemical expression was 200 to 700 times greater in the sustentacular cells of the olfactory neuroepithelium than nasal or tracheal epithelia [12]. In this context, we also examined the immunohistochemical expression of ACE2 using two independent antibodies in 70 head and neck tissues. Interestingly, results obtained were considerably different, in that no labeling was detected by the monoclonal antibody (detecting the ACE-2 extracellular part), unlike the polyclonal antibody (detecting the ACE-2 intracellular part). Using the polyclonal antibody, our immunohistochemical findings were of moderate and expanded staining of epithelial cells, particularly in nasal and oral mucosae, salivary glands and vocal cords, and a weaker distribution in tonsil, pharyngeal and laryngeal supra-glottic epithelia. Since we observed minimal immunohistochemical labeling using the monoclonal antibody, we could hypothesize that the ACE-2 ectodomain was cleaved by ADAM-17. Indeed, Lambert and colleagues have shown that the protein ADAM-17 was responsible for the cleavage of ACE2 [13]. Later, researchers investigated this cleavage in the airway epithelium and confirmed the involvement of this metalloproteinase in the cleavage of ACE2. Remarkably, they have demonstrated that this soluble form remains enzymatically active but partially inhibits the entry of SARS-CoV into cells and therefore limits infection. It is easily assumed that the binding of the virus to the soluble form drastically reduces entry and therefore replication in cells. In addition, they also located the protein cleavage site, namely between amino acids 716 and 741, corresponding to the area targeted by our monoclonal antibody. The cell-associated form of ACE2 (detected by the monoclonal antibody) is, therefore, necessary as a gateway for SARS-CoV-2 [14]. Using the same monoclonal antibody, we were also able to confirm that the neuroepithelium expressed a significantly larger amount of ACE-2 compared to middle turbinate or polyps (Fig. 1). Despite this potential mechanism for epithelial injury, nasal cytological studies in 18 COVID-19 patients, of which 12 reported smell impairment, lacked typical findings of viral induced cellular injury [15], suggesting that direct effects of the virus on the olfactory epithelium cannot likely account for the severity of loss seen in many patients.

CT-Scan Finding of COVID-19 anosmic Patients and ACE-2 immunolabelling of olfactory neuroepithelium and middle turbinate. a Sinus CT-Scan showing completely opacified olfactory cleft in COVID-19 anosmic patient; b Using a ACE-2 monoclonal antibody targeting the extracellular part of the receptor, we were able to show that the neuroepithelium (*) expressed a significantly larger amount of ACE-2 compared to middle turbinate (**)
However, coronaviruses have long been identified as a family of viruses that may be associated with anosmia. In 2007, Suzuki et al. demonstrated that coronavirus may be detected in the nasal discharge of patients with olfactory dysfunction. Moreover, they observed that some patients with normal acoustic rhinometry did not recover their olfaction, suggesting that nasal inflammation and related obstruction were not the only etiological factors underlying the olfactory dysfunction in viral infection [16]. Netland and colleagues have demonstrated on transgenic mice that SARS-CoV may enter the brain through the olfactory bulb, leading to rapid transneuronal spread [17]. Interestingly, authors demonstrated that the virus antigen was first detected 60 to 66 h post-infection and was most abundant in the olfactory bulb. Regions of the cortex (piriform and infralimbic cortices), basal ganglia (ventral pallidum and lateral preoptic regions), and midbrain (dorsal raphe) were also strongly infected after the virus had spread [17]; these regions are connected with the olfactory bulb. The rapid spread of SARS-CoV in the brain was also associated with significant neuronal death.
Emerging autopsy reports have shown SARS-CoV-2 tracking along the olfactory bulb, gyrus rectus and medulla of a patient with COVID related anosmia who subsequently died [18, 19]. More recent imaging reports in the literature show evidence of hyperintense signal and oedema of the olfactory bulb which subsequently resolved [20, 21], giving further support to a central mechanism of anosmia in this group. Previous models of olfactory injury have demonstrated the regenerative capacity of the olfactory epithelium, showing that the basal cells may regenerate and grow new axons back to the olfactory bulb [22]. ‘Rewiring’ of the olfactory bulb, or failures therein may account for problems with discrimination [23]. Scar tissue formation may prevent regrowth, and reducing scar formation using systemic corticosteroids is supported in animal models [24]. Disordered regrowth and a predominance of immature neurons have been shown to be associated with parosmia, which is a common finding amongst patients with Covid-related anosmia [25]. A central mechanism therefore certainly seems to be consistent with the group of patients with more prolonged olfactory deficits. If sustentacular cells seem to be the main SARS-CoV-2 gateway, Chung and colleagues were hypothesized that the infiltration of the olfactory mucosa by CD68 + macrophages expressing SARS-CoV-2 antigens could account for the loss of function [26].
As the pathophysiology of COVID-19 anosmia seems to be better understood, the question of why patients with a moderate to severe form of COVID-19 infection have less olfactory involvement remains unresolved. Yan et al. also found that self-reported olfactory loss associates with milder forms of disease in patients who did not require hospitalization [27]. There is a risk that this represents recall bias due to the increased and over-riding severity of respiratory symptom, and delays in the testing of such patients compared to those with mild disease. If it is indeed true that anosmia is less prevalent in more severe forms, what mechanism may account for this?
Our study comparing mild to severe forms of COVID-19 infection provides some leads. In patients with efficient local immunity, replication of the virus in the nasal and olfactory mucosa may lead to a local inflammatory reaction, involving neuroepithelium and bulb regions (as observed in our brain MRI cases series), and a related otolaryngological pattern of the disease. In that way, we observed that severe patients have less ear, nose and throat complaints than mild patients [2, 3], where the ENT symptoms are prevalent. Considering the hypothesis that patients with a mild form of COVID-19 infection have stronger local immunity, and therefore generate higher rates of local symptoms, conversely patients with severe forms (and with weaker local immunity) would less frequently report ENT symptoms but more respiratory complaints. However, our preliminary results tend to show that patients with a severe or critical forms have higher IgG levels in the serum and nasal secretions than patients with mild form.
Older patients are at higher risk of more severe disease; of course olfactory function diminishes with increasing age [28] and, therefore, hospitalized patients may simply have higher rates of pre-existing loss, and may be less able to detect further decline. Brekenridge and colleagues have described age-related loss of sustentacular cell nuclei as well as olfactory receptor neuron nuclei in animal models [29]. Knowing that sustentacular cells of the neuroepithelium are those which express the most ACE-2, the gateway to SARS-CoV2, one could imagine that the degenerative impairment linked to the age of these cells could explain the less significant incidence anosmia in elderly patients with a severe form of COVID-19 infection. Nasal gene expression of ACE2 was studied on biobank samples taken from individuals aged between 4 and 60 years old, and was found to increase with age; unfortunately, samples from older patients were not available for comparison [30]. As COVID related anosmia appears rare in childhood, and most prevalent in the 40–50 year old age group, perhaps this is related to peak expression in ACE2R and other entry proteins? Of course, this hypothesis calls for further research, but may provide some explanation for the age-related variation in prevalence.
Finally, it has been shown in mouse models that apoptosis of infected olfactory receptor neurons may prevent anterograde propagation of the virus to the olfactory bulb and to the central nervous system [31] Thus, given the capacity to of the olfactory neurones to regenerate, this may be a programmed protective response to neurotropic viruses that lessens the severity of infection, and the lower prevalence of anosmia in more severe disease may reflect a failure of this line of defense.
In summary, the last 3 months have benefited from great international collaborative research, first highlighting and then proving the value of loss of smell and taste as a symptom of COVID-19. Adoption of loss of smell into the case definition by international public health bodies will facilitate control of disease transmission. We have evolved from using self-reported symptoms to incorporating psychophysical testing and imaging into our evaluation and are now able to lend support to hypotheses regarding the underlying etiology. Evaluation of recovery will be ongoing, and we need to better understand how to improve and accelerate recovery. Finally, we need to establish what factors determine the severity of olfactory loss to evaluate whether preventative of olfactory loss is a possibility. There are already over 200 published manuscripts on this subject—we are sure that there will be many more.
References
Hopkins C. Loss of sense of smell as marker of COVID-19 infection https://www.entuk.org/sites/default/files/files/Loss%2520of%2520sense%2520of%2520smell%2520as%2520marker%2520of%2520COVID.pd. Accessed Mar 2020
Lechien JR, Chiesa-Estomba CM, De Siati DR, Horoi M, Le Bon SD, Rodriguez A, Dequanter D, Blecic S, El Afia F, Distinguin L, Chekkoury-Idrissi Y, Hans S, Delgado IL, Calvo-Henriquez C, Lavigne P, Falanga C, Barillari MR, Cammaroto G, Khalife M, Leich P, Souchay C, Rossi C, Journe F, Hsieh J, Edjlali M, Carlier R, Ris L, Lovato A, De Filippis C, Coppee F, Fakhry N, Ayad T, Saussez S (2020) Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): a multicenter European study. Eur Arch Otorhinolaryngol 6:1–11. https://doi.org/10.1007/s00405-020-05965-1
Lechien JR, Chiesa-Estomba CM, Hans S, Barillari MR, Jouffe L, Saussez S (2020) Loss of smell and taste in 2013 European patients with mild to moderate COVID-19. Ann Intern Med. https://doi.org/10.7326/M20-2428
Lechien JR, Cabaraux P, Chiesa-Estomba CM, Khalife M, Plzak J, Hans S, Martiny D, Calvo-Henriquez C, Barillari MR, Hopkins C, Saussez S (2020) Psychophysical olfactory tests and detection of COVID-19 in patients with sudden onset olfactory dysfunction: a prospective study. Ear Nose Throat J 29:145561320929169. https://doi.org/10.1177/0145561320929169
Lechien JR, Cabaraux P, Chiesa-Estomba CM, Khalife M, Hans S, Calvo-Henriquez C, Martiny D, Journe F, Sowerby L, Saussez S (2020) Objective olfactory evaluation of self-reported loss of smell in a case series of 86 COVID-19 patients. Head Neck. https://doi.org/10.1002/hed.26279
Romero-Sánchez CM, Díaz-Maroto I, Fernández-Díaz E et al (2020) Neurologic manifestations in hospitalized patients with COVID-19: the ALBACOVID registry. Neurology. https://doi.org/10.1212/WNL.0000000000009937
Moein ST, Hashemian SMR, Mansourafshar B, Khorram-Tousi A, Tabarsi P, Doty RL (2020) Smell dysfunction: a biomarker for COVID-19. Int Forum Allergy Rhinol. 10(8):944–950. https://doi.org/10.1002/alr.22587
Hornuss D, Lange B, Schroter N, Rieg S, Kern WV, Wagner D (2020) Anosmia in COVID-19 patients. Clin Microbiol Infect. S1198-743X(20):30294–30299. https://doi.org/10.1016/j.cmi.2020.05.017
Vaira LA, Hopkins C, Salzano G, Petrocelli M, Melis A, Cucurullo M et al (2020) Olfactory and gustatory function impairment in COVID-19 patients: Italian objective multicenter-study. Head Neck 42(7):1560–1569
Hopkins C, Surda P, Whithead E, Kumar BN (2020) Early recovery following new onset anosmia during the COVID-19 pandemic: an observational cohort study. J Otolaryngol Head Neck Surg 49(1):26. https://doi.org/10.1186/s40463-020-00423-8
Vaira L, Hopkins C, Petrocelli M, Lechien J, Chiesa-Estomaba C et al (2020) Smell and taste recovery in COVID-19 patients: A 60 day objective and prospective study. J Laryngol Otol. https://doi.org/10.1017/S0022215120001826
Chen M, Shen W, Rowan N et al (2020) Elevated ACE2 expression in the olfactory neuroepithelium: implications for anosmia and upper respiratory SARS-COV-2 entry and replication. BioRxiv. https://doi.org/10.1101/2020.05.08.084996
Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI et al (2005) Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem 280(34):30113–30119
Jia HP, Look DC, Tan P, Shi L, Hickey M, Gakhar L et al (2009) Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol 297(1):L84–96
Gelardi M, Notargiacomo M, Trecca EMC, Cassano M, Ciprandi G (2020) COVID-19 and nasal cytobrush cytology. Acta Cytol 64(4):397–398
Suzuki M, Saito K, Min WP, Vladau C, Toida K, Itoh H, Murakami S (2007) Identification of viruses in patients with postviral olfactory dysfunction. Laryngoscope 117(2):272–277
Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S (2008) Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol 82(15):7264–7275. https://doi.org/10.1128/JVI.00737-08
Bulfamante G, Chiumello D, Canevini MP et al (2020) First ultrastructural autoptic findings of SARS-Cov-2 in olfactory pathways and brainstem. Min Anestesiol. 86(6):678–679. https://doi.org/10.23736/S0375-9393.20.14772-2
Meinhardt J, Radke J, Dittmayer C et al. Olfactory Transmucosal SARS-Cov-2 Invasion as Port Of Central Nervous System Entry In COVID-19 Patients. 2020; bioRxiv preprint. https://www.biorxiv.org/content/10.1101/2020.06.04.135012v1
Laurendon T, Radulesco MJ et al (2020) Bilateral transient olfactory bulbs edema during COVID-19-related anosmia. Neurology. https://doi.org/10.1212/WNL.0000000000009850
Politi LS, Salsano E, Grimaldi M (2020) Magnetic resonance imaging alteration of the brain in a patient with coronavirus disease 2019 (COVID-19) and anosmia. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2020.2125(Published online May 29, 2020)
Graziadei PPC, Karlan MS, Monti Graziadei GA, Bernstein JJ (1980) Neurogenesis of sensory neurons in the primate olfactory system after section of the fila olfactoria. Brain Res 186:289–300
Schwob JE (2002) Neural regeneration and the peripheral olfactory system. Anat Rec 269(1):33–49
Tian J, Pinto JM, Xin Y et al (2015) Dexamethasone affects mouse olfactory mucosa gene expression and attenuates genes related to neurite outgrowth. Int Forum Allergy Rhinol 5(10):907–918. https://doi.org/10.1002/alr.21586Epub 2015 Jul 3
Leopold DA, Loehrl TA, Schwob JE (2002) Long-term follow-up of surgically treated phantosmia. Arch Otolaryngol Head Neck Surg 128642:647
Chung TW, Sridhar TW, Zhang AJ (2020) Olfactory dysfunction in coronavirus disease 2019 patients: observational cohort study and systematic review. Open Forum Infect Dis 7(6):ofaa199
Yan CH, Faraji F, Prajapati DP et al (2020) Self-reported olfactory loss associates with outpatient clinical course in COVID-19. Int Forum Allergy Rhinol. https://doi.org/10.1002/alr.22592
Ship JA, Pearson JD, Cruise LJ, Brant LJ, Metter EJ (1996) Longitudinal changes in smell identification. Journal of gerontology series A. Biol Sci Med Sci 51:86–91
Breckenridge LJ, Cameron J, Puri N et al (1997) Localised degeneration occurs in aged mouse olfactory epithelium. J Anat 191:151–154. https://doi.org/10.1046/j.1469-7580.1997.19110151
Bunyavanich S, Do A, Vicencio A (2020) Nasal gene expression of angiotensin-converting enzyme 2 in children and adults. JAMA 323(23):2427–2429. https://doi.org/10.1001/jama.2020.8707
van Riel D, Verdijk R, Kuiken T (2015) The olfactory nerve: a shortcut for influenza and other viral diseases into the central nervous system. J Pathol 235(2):277–287
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Saussez, S., Lechien, J.R. & Hopkins, C. Anosmia: an evolution of our understanding of its importance in COVID-19 and what questions remain to be answered. Eur Arch Otorhinolaryngol 278, 2187–2191 (2021). https://doi.org/10.1007/s00405-020-06285-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00405-020-06285-0