
Abstract
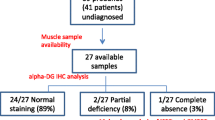
Protein O-glucosyltransferase 1 (POGLUT1) activity is critical for the Notch signaling pathway, being one of the main enzymes responsible for the glycosylation of the extracellular domain of Notch receptors. A biallelic mutation in the POGLUT1 gene has been reported in one family as the cause of an adult-onset limb-girdle muscular dystrophy (LGMD R21; OMIM# 617232). As the result of a collaborative international effort, we have identified the first cohort of 15 patients with LGMD R21, from nine unrelated families coming from different countries, providing a reliable phenotype–genotype and mechanistic insight. Patients carrying novel mutations in POGLUT1 all displayed a clinical picture of limb-girdle muscle weakness. However, the age at onset was broadened from adult to congenital and infantile onset. Moreover, we now report that the unique muscle imaging pattern of “inside-to-outside” fatty degeneration observed in the original cases is indeed a defining feature of POGLUT1 muscular dystrophy. Experiments on muscle biopsies from patients revealed a remarkable and consistent decrease in the level of the NOTCH1 intracellular domain, reduction of the pool of satellite cells (SC), and evidence of α-dystroglycan hypoglycosylation. In vitro biochemical and cell-based assays suggested a pathogenic role of the novel POGLUT1 mutations, leading to reduced enzymatic activity and/or protein stability. The association between the POGLUT1 variants and the muscular phenotype was established by in vivo experiments analyzing the indirect flight muscle development in transgenic Drosophila, showing that the human POGLUT1 mutations reduced its myogenic activity. In line with the well-known role of the Notch pathway in the homeostasis of SC and muscle regeneration, SC-derived myoblasts from patients’ muscle samples showed decreased proliferation and facilitated differentiation. Together, these observations suggest that alterations in SC biology caused by reduced Notch1 signaling result in muscular dystrophy in LGMD R21 patients, likely with additional contribution from α-dystroglycan hypoglycosylation. This study settles the muscular clinical phenotype linked to POGLUT1 mutations and establishes the pathogenic mechanism underlying this muscle disorder. The description of a specific imaging pattern of fatty degeneration and muscle pathology with a decrease of α-dystroglycan glycosylation provides excellent tools which will help diagnose and follow up LGMD R21 patients.







Similar content being viewed by others

References
Acar M, Jafar-Nejad H, Takeuchi H, Rajan A, Ibrani D, Rana NA et al (2008) Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell 132:247–258. https://doi.org/10.1016/j.cell.2007.12.016
Alonso-Jimenez A, Kroon R, Alejaldre-Monforte A, Nunez-Peralta C, Horlings CGC, van Engelen BGM et al (2019) Muscle MRI in a large cohort of patients with oculopharyngeal muscular dystrophy. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2018-319578
Balcin H, Palmio J, Penttila S, Nennesmo I, Lindfors M, Solders G et al (2017) Late-onset limb-girdle muscular dystrophy caused by GMPPB mutations. Neuromusc Disord NMD 27:627–630. https://doi.org/10.1016/j.nmd.2017.04.006
Bankole LC, Feasson L, Ponsot E, Kadi F (2013) Fibre type-specific satellite cell content in two models of muscle disease. Histopathology 63:826–832. https://doi.org/10.1111/his.12231
Bischof J, Maeda RK, Hediger M, Karch F, Basler K (2007) An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci USA 104:3312–3317
Bjornson CR, Cheung TH, Liu L, Tripathi PV, Steeper KM, Rando TA (2012) Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 30:232–242. https://doi.org/10.1002/stem.773
Bonne G, Rivier F, Hamroun D (2018) The 2019 version of the gene table of neuromuscular disorders (nuclear genome). NMD 28:1031–1063. https://doi.org/10.1016/j.nmd.2018.09.006
Bonnemann CG (2011) The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol 7:379–390. https://doi.org/10.1038/nrneurol.2011.81
Brohl D, Vasyutina E, Czajkowski MT, Griger J, Rassek C, Rahn HP et al (2012) Colonization of the satellite cell niche by skeletal muscle progenitor cells depends on Notch signals. Dev Cell 23:469–481. https://doi.org/10.1016/j.devcel.2012.07.014
Cirak S, Foley AR, Herrmann R, Willer T, Yau S, Stevens E et al (2013) ISPD gene mutations are a common cause of congenital and limb-girdle muscular dystrophies. Brain J Neurol 136:269–281. https://doi.org/10.1093/brain/aws312
Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA et al (2005) Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 122:289–301. https://doi.org/10.1016/j.cell.2005.05.010
Diaz-Manera J, Fernandez-Torron R, James MK, Mayhew A, Smith FE, Moore UR et al (2018) Muscle MRI in patients with dysferlinopathy: pattern recognition and implications for clinical trials. J Neurol Neurosurg Psychiatry 89:1071–1081. https://doi.org/10.1136/jnnp-2017-317488
Fernandez-Valdivia R, Takeuchi H, Samarghandi A, Lopez M, Leonardi J, Haltiwanger RS et al (2011) Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development 138:1925–1934. https://doi.org/10.1242/dev.060020
Figueroa-Bonaparte S, Llauger J, Segovia S, Belmonte I, Pedrosa I, Montiel E et al (2018) Quantitative muscle MRI to follow up late onset Pompe patients: a prospective study. Sci Rep 8:10898. https://doi.org/10.1038/s41598-018-29170-7
Fischer D, Walter MC, Kesper K, Petersen JA, Aurino S, Nigro V et al (2005) Diagnostic value of muscle MRI in differentiating LGMD2I from other LGMDs. J Neurol 252:538–547. https://doi.org/10.1007/s00415-005-0684-4
Gildor B, Schejter ED, Shilo BZ (2012) Bidirectional Notch activation represses fusion competence in swarming adult Drosophila myoblasts. Development 139:4040–4050. https://doi.org/10.1242/dev.077495
Goddeeris MM, Wu B, Venzke D, Yoshida-Moriguchi T, Saito F, Matsumura K et al (2013) LARGE glycans on dystroglycan function as a tunable matrix scaffold to prevent dystrophy. Nature 503:136–140. https://doi.org/10.1038/nature12605
Harvey BM, Haltiwanger RS (2018) Regulation of Notch function by O-glycosylation. Adv Exp Med Biol 1066:59–78. https://doi.org/10.1007/978-3-319-89512-3_4
Jiang C, Wen Y, Kuroda K, Hannon K, Rudnicki MA, Kuang S (2014) Notch signaling deficiency underlies age-dependent depletion of satellite cells in muscular dystrophy. Disease Models Mech 7:997–1004. https://doi.org/10.1242/dmm.015917
Kottlors M, Kirschner J (2010) Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res 340:541–548. https://doi.org/10.1007/s00441-010-0976-6
Kuang S, Kuroda K, Le Grand F, Rudnicki MA (2007) Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 129:999–1010. https://doi.org/10.1016/j.cell.2007.03.044
Li Z, Fischer M, Satkunarajah M, Zhou D, Withers SG, Rini JM (2017) Structural basis of Notch O-glucosylation and O-xylosylation by mammalian protein-O-glucosyltransferase 1 (POGLUT1). Nat Commun 8:185. https://doi.org/10.1038/s41467-017-00255-7
Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C et al (1998) Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 20:31–36. https://doi.org/10.1038/1682
Mercuri E, Lampe A, Allsop J, Knight R, Pane M, Kinali M et al (2005) Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. NMD 15:303–310. https://doi.org/10.1016/j.nmd.2005.01.004
Mercuri E, Pichiecchio A, Allsop J, Messina S, Pane M, Muntoni F (2007) Muscle MRI in inherited neuromuscular disorders: past, present, and future. JMRI 25:433–440. https://doi.org/10.1002/jmri.20804
Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS et al (2002) Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 418:417–422. https://doi.org/10.1038/nature00837
Muntoni F, Torelli S, Wells DJ, Brown SC (2011) Muscular dystrophies due to glycosylation defects: diagnosis and therapeutic strategies. Curr Opin Neurol 24:437–442. https://doi.org/10.1097/WCO.0b013e32834a95e3
Paradas C, Gonzalez-Quereda L, De Luna N, Gallardo E, Garcia-Consuegra I, Gomez H et al (2009) A new phenotype of dysferlinopathy with congenital onset. NMD 19:21–25. https://doi.org/10.1016/j.nmd.2008.09.015
Rahimov F, Kunkel LM (2013) The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol 201:499–510. https://doi.org/10.1083/jcb.201212142
Ralser DJ, Takeuchi H, Fritz G, Basmanav FB, Effern M, Sivalingam S et al (2019) Altered Notch signaling in Dowling-Degos disease: additional mutations in POGLUT1 and further insights into disease pathogenesis. J Invest Dermatol 139:960–964. https://doi.org/10.1016/j.jid.2018.10.030
Rana NA, Nita-Lazar A, Takeuchi H, Kakuda S, Luther KB, Haltiwanger RS (2011) O-glucose trisaccharide is present at high but variable stoichiometry at multiple sites on mouse Notch1. J Biol Chem 286:31623–31637. https://doi.org/10.1074/jbc.M111.268243
Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM (2008) Self-renewal and expansion of single transplanted muscle stem cells. Nature 456:502–506. https://doi.org/10.1038/nature07384
Sacco A, Mourkioti F, Tran R, Choi J, Llewellyn M, Kraft P et al (2010) Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 143:1059–1071. https://doi.org/10.1016/j.cell.2010.11.039
Servian-Morilla E, Cabrera-Serrano M, Rivas-Infante E, Carvajal A, Lamont PJ, Pelayo-Negro AL et al (2019) Altered myogenesis and premature senescence underlie human TRIM32-related myopathy. Acta Neuropathol Commun 7:30. https://doi.org/10.1186/s40478-019-0683-9
Servian-Morilla E, Takeuchi H, Lee TV, Clarimon J, Mavillard F, Area-Gomez E et al (2016) A POGLUT1 mutation causes a muscular dystrophy with reduced Notch signaling and satellite cell loss. EMBO Mol Med 8:1289–1309. https://doi.org/10.15252/emmm.201505815
Sewry CA, Taylor J, Anderson LV, Ozawa E, Pogue R, Piccolo F et al (1996) Abnormalities in alpha-, beta- and gamma-sarcoglycan in patients with limb-girdle muscular dystrophy. NMD 6:467–474
Sheikh MO, Halmo SM, Patel S, Middleton D, Takeuchi H, Schafer CM et al (2017) Rapid screening of sugar-nucleotide donor specificities of putative glycosyltransferases. Glycobiology 27:206–212. https://doi.org/10.1093/glycob/cww114
Straub V, Murphy A, Udd B (2018) 229th ENMC international workshop: Limb girdle muscular dystrophies—nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. NMD 28:702–710. https://doi.org/10.1016/j.nmd.2018.05.007
Takeuchi H, Yu H, Hao H, Takeuchi M, Ito A, Li H et al (2017) O-Glycosylation modulates the stability of epidermal growth factor-like repeats and thereby regulates Notch trafficking. J Biol Chem 292:15964–15973. https://doi.org/10.1074/jbc.M117.800102
Varshney S, Stanley P (2018) Multiple roles for O-glycans in Notch signalling. FEBS Lett 592:3819–3834. https://doi.org/10.1002/1873-3468.13251
Venken KJ, He Y, Hoskins RA, Bellen HJ (2006) P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 314:1747–1751
Wattjes MP, Kley RA, Fischer D (2010) Neuromuscular imaging in inherited muscle diseases. Eur Radiol 20:2447–2460. https://doi.org/10.1007/s00330-010-1799-2
Willer T, Lee H, Lommel M, Yoshida-Moriguchi T, de Bernabe DB, Venzke D et al (2012) ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat Genet 44:575–580. https://doi.org/10.1038/ng.2252
Yu H, Takeuchi H (2019) Protein O-glucosylation: another essential role of glucose in biology. Curr Opin Struct Biol 56:64–71. https://doi.org/10.1016/j.sbi.2018.12.001
Yu H, Takeuchi H, Takeuchi M, Liu Q, Kantharia J, Haltiwanger RS et al (2016) Structural analysis of Notch-regulating Rumi reveals basis for pathogenic mutations. Nat Chem Biol 12:735–740. https://doi.org/10.1038/nchembio.2135
Acknowledgements
We thank Stephan Kröger (Münich University) for kind donation of the antibody against the α-dystroglycan core (clone no. 317); Developmental Studies Hybridoma Bank for the 22C10 antibody; Bloomington Drosophila stock center (NIH P40OD018537) for fly strains; and Confocal Microscopy Core of the BCM IDDRC (1U54HD083092; the Eunice Kennedy NICHD) and Sandra Donkervoort (NINDS/NIH) for help with genetic studies.
Funding
This work was supported in part by the Instituto de Salud Carlos III and FEDER (FIS PI16-01843 to C. Paradas and JR15/00042 to M. Cabrera-Serrano), the Consejería de Salud, Junta de Andalucía (PI-0085-2016 and PE-S1275 to C. Paradas, and B-0005-2017 to M. Cabrera-Serrano), NIH/NIGMS (R01GM084135 and R35GM130317 to H. Jafar-Nejad, and R01GM061126 to R.S. Haltiwanger), JSPS KAKENHI Grants-in-Aid for Research Activity Start-up and Scientific Research (B) (JP17H06743 and JP19H03176 to H. Takeuchi), and Takeda Science Foundation and Daiichi Sankyo Foundation of Life Science (to H. Takeuchi). MYO-SEQ has been supported by Sanofi Genzyme, Ultragenyx, the LGMD2I Research Fund, Samantha J Brazzo Foundation, LGMD2D Foundation, Kurt + Peter Foundation, Muscular Dystrophy UK and Coalition to Cure Calpain 3. Work in CGB’s group is supported by NINDS/NIH intramural funds.
Author information
Authors and Affiliations
Contributions
CP and ES-M designed the study. ES-M performed α-dystroglycan expression and function studies, cell culture, satellite cells and myogenesis analysis. ER and ES-M processed and studied muscle biopsies. MC, KJ, AT, OA, and VS analyzed the genetic studies. CP, MC, TCh, AT, NM, TM, SN, KC, RG, CB, LM, JB, JV, and IT clinically identified and characterized patients and collected muscle MRI and muscle samples. AI, MT, HH, HT, and RSH designed, analyzed and performed biochemical in vitro assays. AP and HJN designed, analyzed and performed Drosophila experiments. CP supervised and coordinated all work. CP, ES-M, HJN, RSH, HT, and AP contributed to preparation of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared that no conflict of interest exists.
Ethics approval and consent to participate
This study was approved by the Institutional Research Ethic Committee at Hospital Universitario Virgen del Rocío in Sevilla (Spain). Written informed consent was received from participants, prior to inclusion in the study, for genetic studies, for muscle biopsies, and for pictures appearing in the manuscript.
Consent for publication
All authors consented to publication of this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Servián-Morilla, E., Cabrera-Serrano, M., Johnson, K. et al. POGLUT1 biallelic mutations cause myopathy with reduced satellite cells, α-dystroglycan hypoglycosylation and a distinctive radiological pattern. Acta Neuropathol 139, 565–582 (2020). https://doi.org/10.1007/s00401-019-02117-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-019-02117-6