
Abstract
Tumours can be viewed as aberrant tissues or organs sustained by tumorigenic stem-like cells that engage into dysregulated histo/organogenetic processes. Paragangliomas, prototypical organoid tumours constituted by dysmorphic variants of the vascular and neural tissues found in normal paraganglia, provide a model to test this hypothesis. To understand the origin of paragangliomas, we built a biobank comprising 77 cases, 18 primary cultures, 4 derived cell lines, 80 patient-derived xenografts and 11 cell-derived xenografts. We comparatively investigated these unique complementary materials using morphofunctional, ultrastructural and flow cytometric assays accompanied by microRNA studies. We found that paragangliomas contain stem-like cells with hybrid mesenchymal/vasculoneural phenotype, stabilized and expanded in the derived cultures. The viability and growth of such cultures depended on the downregulation of the miR-200 and miR-34 families, which allowed high PDGFRA and ZEB1 protein expression levels. Both tumour tissue- and cell culture-derived xenografts recapitulated the vasculoneural paraganglioma structure and arose from mesenchymal-like cells through a fixed developmental sequence. First, vasculoangiogenesis organized the microenvironment, building a perivascular niche which in turn supported neurogenesis. Neuroepithelial differentiation was associated with severe mitochondrial dysfunction, not present in cultured paraganglioma cells, but acquired in vivo during xenograft formation. Vasculogenesis was the Achilles’ heel of xenograft development. In fact, imatinib, that targets endothelial-mural signalling, blocked paraganglioma xenograft formation (11 xenografts from 12 cell transplants in the control group versus 2 out of 10 in the treated group, P = 0.0015). Overall our key results were unaffected by the SDHx gene carrier status of the patient, characterized for 70 out of 77 cases. In conclusion, we explain the biphasic vasculoneural structure of paragangliomas and identify an early and pharmacologically actionable phase of paraganglioma organization.
Similar content being viewed by others


Introduction
The parallelism between some tumours and their organs or tissues of origin suggests that tumourigenesis, like organogenesis, might pass through definable formative stages, orchestrated by stem-like cells whose engagement in complementary, albeit aberrant, differentiation processes could be spatiotemporally connected with the organization of the neoplastic microenvironment [15, 30]. This hypothesis implies that preventive and curative interventions should be designed and targeted based on a thorough understanding of tumour histogenesis.
Paragangliomas (PGLs) are rare hypervascular tumours of the paraganglia, organelles connected with the autonomic branches of the cranial (head and neck) and thoraco-lumbar nerves [47]. They are frequently associated with predisposing mutations in one of several nuclear genes, most relevantly SDHA/B/C/D, that encode the four subunits of succinate dehydrogenase (SDH), a mitochondrial (mt) enzyme participating in both oxidative phosphorylation and the Krebs cycle, and SDHAF2, whose product is required for SDHA flavination (collectively the SDHx genes) [47]. PGLs grow slowly, but are highly infiltrating, may unpredictably metastasize and are refractory to chemo/radiotherapy. Head and neck PGLs (~ 20% of all PGLs) are of particular concern, as they spread along the regional neurovascular structures towards the skull base, may insinuate intracranially and may compress the brainstem [61]. Surgical resection is challenging, and postoperative deficits of the lower cranial nerves are a significant cause of morbidity and permanent disability [4]. The difficult recruitment of patients, the need of long follow-up and the lack of preclinical models are major barriers to the development or repurposing of drugs for PGL treatment [47, 61].
PGLs recapitulate the histostructure of normal paraganglia. The cardinal feature shared by PGLs and paraganglia is the integration of a neurosecretory network, consisting in nests or cords of glia-bound neuroepithelial cells (“zellballens”), with an angiomatous vasculature [7]. The histostructural convergence suggests that paragangliar tumorigenesis exploits a deeply embedded organogenetic program. In this regard stem-like cells have been detected in PGLs [9, 46, 75]. However, the current view, reflected in the WHO classification [71], is that PGLs are of neuroendocrine (i.e., neuroepithelial) origin, while their vasculature, although aberrant, is thought to arise from extrinsic angiogenic ingrowth and is thus relegated to a secondary and subordinate role [40]. This influences the current strategies of PGL prevention and therapy [47, 61].
Here, using SDHx-characterized head and neck PGLs and derived in vitro and in vivo models, we demonstrate that the vascular and the neural PGL components develop sequentially from stem-like tumour cells characterized by a hybrid mesenchymal/vasculoneural phenotype. Thus, PGLs are fundamentally vasculoneural tumours and vasculoangiogenesis is the primary and pharmacologically actionable tumorigenic process. Our findings, that support an organogenetic model of paragangliar tumourigenesis, are applicable to PGLs that are both related and unrelated to germline SDHx mutations.
Patients, materials, and methods
Patients, samples and SDHx mutational analysis
The case series (77 PGL patients recruited between November 2009 and June 2017 at Gruppo Otologico, Piacenza, Italy) is listed in Table S1 (Online Resource 1). The patients did not receive radio/chemotherapy but preoperative tumour embolization was routinely performed (except for patients with tympanic PGL) [61]. Case acronyms encode PGL (P) localization (carotid body, C; vagal, V; tympanic, T; tympano-jugular, TJ) followed by progressive number. Solid biospecimens, evaluated fresh to exclude areas damaged by embolization, were differentially sampled within 5 min from excision in: (a) RNAlater (nucleic acids); (b) high-glucose DMEM with penicillin, streptomycin and fungizone (cytofluorimetry, cell culture, ex vivo culture, xenotransplantation, JC-1 assays); (c) liquid nitrogen (biochemical studies); (d) 2% paraformaldehyde (PFA) and 0.2% glutaraldehyde in PBS at 4 °C (8 h), then 2% PFA (ApoTome immunofluorescence, AIF); (e) 2% glutaraldehyde in PBS at 4 °C (light and transmission electron microscopy, TEM). Samples (d)–(e) were trimmed in ~ 3 × 3 mm pieces before fixation. Processing was restricted to (c)–(e) when scarce tissue was available. Anticoagulated blood (20 ml) for mutational analysis and formalin-fixed/paraffin-embedded (FFPE) samples for standard histopathology and immunohistochemistry (IHC) were routinely obtained.
Point mutations and large deletions/rearrangements in the SDHA, SDHB, SDHC, SDHD and SDHAF2 genes and SDHB protein immunostaining were assessed as described [7, 67]. Methods used for miRNAstudies are detailed in the Online Resource 2 (Appendix to Materials and Methods).
Immunomorphological and ultrastructural studies
AIF, that generates high resolution images in the focal plane by computational optical sectioning [57], was used to investigate the expression and localization of marker proteins in semithin (200 nm-thick) cryo-ultramicrotomy sections of PGL and patient-derived xenograft (PDX) tissues that had been lightly fixed in 2% PFA and 0.2% glutaraldehyde in cold PBS at surgery (see above). Sections were analysed using an ApoTome Axio Observer Z1 inverted microscope (Zeiss) equipped with an AxioCam MRm Rev.3. Colocalizations were assessed with Axio Vision software, release 4.6.3 (Zeiss). AIF was also used to analyse cells, which were grown on CultureSlides (BD Falcon), fixed in 4% PFA for 30 min, washed twice in PBS, stained and directly examined as above. Alternatively, cells were analysed using an LSM 800 confocal laser scanning microscope (Zeiss). Marker proteins and relative antibodies are listed in the Online Resource 2 (Appendix to Materials and Methods). Positive and negative controls were added to all the immunostaining procedures. Negative controls included omission of primary antibody, blocking with secondary antibody and substitution of primary antibody with irrelevant isotype-matched immunoglobulin. Standard IHC on FFPE sections for S100, synaptophysin (SYP), chromogranin A (CGA), vimentin, alpha-smooth muscle actin (SMA), CD34 and NOTCH1 was as reported [7]. For TEM, PGL, PDX and CDX samples (~ 3 mm in maximum diameter) or cell pellets (at least 1 × 106 cells) were processed as described [7] and examined using a Philips CM10 transmission electron microscope. Mitochondrial cross-sectional areas (µm2) were determined on electron micrographs using NIH ImageJ and analysed with one-way ANOVA and paired samples t test. Methods used for cytofluorimetry and western blotting are detailed in the Online Resource 2.
Cell cultures
Primary PGL cultures were established from cells dissociated with Accumax (Millipore) according to manufacturer’s protocol and grown at 37 °C under ambient atmosphere plus 5% CO2 in DMEM-F12 supplemented with 20–10% FBS (early to late passages), 3.5 mM l-glutamine, 100 IU penicillin and 100 μg/ml streptomycin. Primary cultures were immortalized by retroviral-mediated transduction of full-length hTERT and/or simian virus 40 (SV40) large-tumour (LT) antigen (Addgene, https://www.addgene.org/). The virus was constructed infecting HEK293 cells with a combination of vectors bringing Gag-polymerase, virus envelope proteins and full-length hTERT (pBabe-hygro-hTERT) or SV40LT (pBabe-puro SV40 LT) cDNAs. Mid-confluence cultures at passage 4 were exposed to 0.4 µm filtered supernatants from the retroviral packaging lines for 3–6 h with 5 µg/ml polybrene, and grown for two passages before second infection (when performed). Cells were selected with puromycin and/or hygromycin B until all untransduced cells had been eliminated.
For neurosphere formation primary or immortalized cells were cultured serum-free in CellRepel flasks (Eppendorf) with DMEM:F12 or NeuroBasal media supplemented with N2, B27 (Life Technologies) and growth factors (FGFb, IGF1, EGF, each 20 ng/ml) (Prospec). For the Matrigel assay [19] cells (9 × 104/well) were seeded in triplicate in 35 mm Cell Imaging Dishes (Eppendorf) on top of a layer of growth factor-reduced Matrigel (BD) in DMEM:M199 (1:1) supplemented with 20% FBS, 18U/ml heparine (Clarisco) and 50 μg/ml endothelial cell growth factor (Sigma). The cultures were examined by phase contrast at 1–2–4 h, fixed for 30 min adding 4% PFA in PBS to an equal volume of medium, washed twice in PBS and permeabilized with NET-gel (5 mM EDTA, 50 mM Tris–HCl pH 7.4, 0.05% NP-40, 0.25% carragenin lambda gelatin; 0.02% NaN3) for 40 min. The short incubation time, quite comparable to that used for HUVEC cells in some studies (6 h) [10], best suited the rapid self-organization of PGL cells into a web-like network. Primary antibodies to CD34 (Ancell mouse monoclonal #183-040), CD31 (BD Pharmigen mouse monoclonal #555445), DLK1 (Abcam mouse monoclonal #119930) and platelet-derived growth factor receptor alpha (PDGFRA, Santa Cruz rabbit polyclonal sc-338) were diluted in NetGel (1:50 except anti-DLK1, 1:500) and incubated overnight. After washing twice with NetGel, goat anti-mouse (Invitrogen #A11017) or goat anti-rabbit (Invitrogen #A11070), both diluted 1:100 in NetGel, were incubated for 1 h, washed once with NetGel and once with PBS and examined with an LSM 510 confocal microscope (Zeiss). The neuroblastoma cell line SH-SY5Y (ATCC; CRL-2266), acquired in 2007 and authenticated in January 2013 [7], was cultured in RPMI 1640 (GE Healthcare) containing 10% FBS, 2 mM l-glutamine, 100 IU penicillin, 100 μg/ml streptomycin [7]. Control human embryo lung fibroblast MRC5 cells, obtained from Karolinska Institutet, Stockholm, were cultivated in eMEM containing 10% FBS and 2 mM l-glutamine, 100 IU penicillin, 100 μg/ml streptomycin.
Ex vivo cultures and xenografts
For ex vivo culture PGL samples taken at surgery in high-glucose DMEM with penicillin, streptomycin and fungizone were cut into ~ 3 × 3 mm cubes under sterile conditions and cultured at 37 °C in 12-well plates (as used for cells) for up to 10 days. No enzymatic dissociation was used. Samples were then processed and analysed as described above. To obtain PDXs, fresh PGL tissues, sampled and cut as above, were transplanted subcutaneously within 24 h from surgery into the flank(s) and/or under the neck skinfold(s) of 6–12 weeks old NOD scid gamma (NSG) mice (when possible sex-matched to the donor patient). Normal tissues (abdominal skin) from PGL patients were transplanted as controls. The immortalized PTJ64i PGL cell line, that can be grown in large quantities, was used to obtain cell-derived xenografts (CDXs). To this end athymic female CD-1 nude mice aged 4 weeks were acquired from Charles River and, after 2 weeks of quarantine, grafted in both flanks with 9 × 106 PTJ64i cells in 0.1 ml PBS. All mice were maintained in temperature-controlled cages docked on a ventilated rack with HEPA filters, euthanized by CO2 inhalation followed by exsanguination via cardiac puncture and, in some cases, intracardiac perfusion with 1 ml India ink solution (1/20 in sterile H2O with 10 IU/ml heparin), to evaluate the integration between PDX and murine vasculature. Mice were finally dissected to identify putative growths in transplant areas, that were measured, photographed and, depending on tissue availability, sampled as described for PGLs. The thoracoabdominal organs were routinely examined and sampled for standard microscopy.
Mt DNA analysis
DNAs from PDXs, blood of the donor patients and host mouse tissues were extracted using the PureLink Genomic DNA Kit (Invitrogen) and quantified with Nanodrop 2000 (Thermo Scientific). HVR1 mtDNA typing was performed via PCR and direct automated sequencing using primers L15990 (sense) and H16434 (antisense) [56]. Sequences were aligned and compared to the HVR1 consensus reference sequence.
JC-1 staining
The cell-permeant vital dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide, Invitrogen) is concentrated in mitochondria with functional OXPHOS. This results in a fluorescence emission shift from green (~ 529 nm) to red (~ 590 nm), due to concentration-dependent formation of intramitochondrial J-aggregates that accurately reflect the mitochondrial membrane potential (mt ΔΨ) [52]. For JC-1 staining PGL tissues and autologous controls (subcutaneous white adipose tissue, WAT) were obtained at surgery, maintained in high-glucose DMEM with penicillin, streptomycin and fungizone for up to 24 h, minced aseptically in ~ 3 × 3 mm fragments, incubated for 10 min at 37 °C in culture medium containing 15% FBS and 5 µM JC-1, washed for 1 min with warm PBS and placed in Cell Imaging Dishes (Eppendorf). Then the red/green fluorescence intensity ratios, that are specific indicators of mt ΔΨ [52], were independently calculated for the vascular/glial and the neuroepithelial PGL components and compared to the red/green fluorescence ratios calculated for the WAT controls. PGL cell cultures and control MRC5 cells, live on CultureSlides (BD Falcon), were JC-1 stained in PBS plus 5 µM DRAQ5 (Thermo Fisher Scientific) to counterstain nuclei. Tissues and cells were analysed with an LSM 510 confocal microscope (Zeiss).
Imatinib treatments
Stock solutions of imatinib (Selleck Chemicals) in sterile H2O (200 mM) were stored at − 20 °C. The imatinib concentration for in vitro treatment (10 μM) was chosen based on IC50 values determined on PGL cell cultures using the Alamar Blue cell viability assay (Invitrogen) and the trypan blue exclusion method [18]. Imatinib or an equal volume of sterile H2O were added to triplicate wells of 24-well plates seeded with 5000 cells. Cells were incubated in a humidified chamber at 37 °C-5% CO2 with imatinib- or H2O-supplemented medium replaced every 24 h and counted at 24, 48 and 72 h using the trypan blue exclusion method. Significance was determined by two-way ANOVA with 95% CI and Bonferroni correction, statistical analyses were performed with Prism 6 (GraphPad Software, San Diego, CA). Caspase activity (average of triplicates normalized to controls) was evaluated with the Caspase-Glo 3/7® Assay (Promega) using a Veritas microplate luminometer (Turner BioSystems). Significance was calculated with the unpaired t test. Treated and control cultures were analysed by AIF and WB for ZEB1 and PDGFRA, by AIF for BAX, and by WB for antiphosphotyrosine, PDGFRB, Beclin 1, vinculin, and β-actin. Antibodies were as reported above. MicroRNA levels before and after treatment were evaluated as described [7].
In vivo prevention of CDX formation by imatinib was evaluated on 11 female CD-1 mice (Charles River) grafted in both flanks with 9 × 106 PTJ64i cells in 0.1 ml PBS at 6 weeks of age. The mice were randomly assigned to two groups and injected i.p. once daily, starting day 3, with either a preselected bulk dose of imatinib in sterile H2O (50 mg/Kg, Biorbyt, Cambridge, UK), adapted from the literature [13] (5 mice, 10 grafts), or an equal volume of sterile H2O (6 mice, 12 grafts), for 20 days. Treatment was then scaled-down to a one-third dose (16.6 mg/Kg) for 20 additional days, after which the mice were euthanized, analysed and sampled as above. Body weight was measured weekly throughout treatment. Autopsy and standard microscopic analysis of the thoraco-abdominal organs were routinely performed.
Results
Characteristics of the tumours
Background information on the examined series of 77 head and neck PGL cases, including SDHx mutation carrier status of the patients and tumour immunostaining for the SDHB protein, surrogate marker of SDHx mutations [24], are described in the Online Resource 1 (Tables S1–S3). All the tumours were immunophenotyped using standard IHC on paraffin sections and AIF on semithin cryosections. The fine morphology was routinely investigated by TEM.
By IHC markers associated with endothelial (CD34), pericytic (SMA), glial (S100) and neuroepithelial (SYP) differentiation clearly discriminated the four major cell types that define the PGL microenvironment, but we observed that these otherwise phenotypically distinct cell types coexpressed to variable extents the mesenchymal markers vimentin and NOTCH1 (Online Resource 2, Fig. S1). AIF analyses confirmed that PGL cells of vascular and neural lineage always coexpressed immature markers associated with mesenchymal stem cells (CD44, vimentin) [14, 74, 75], the hypoxia-ZEB1 axis (HIF2A, ZEB1, NOTCH1, DLK1) [33, 37, 41, 42, 59, 70, 77], glucose uptake (GLUT4) [59] and vasculoneurogenesis (NOTCH1, DLK1, nestin, PDGFRA, KIT/CD117, VEGFR1, VEGFR2) [9, 11, 31, 38, 64] (Table 1, Fig. 1). Other markers were lineage-specific, including β2-microglobulin, CD34 and CD31 for endothelial cells, SMA for pericytes; S100 and GFAP for glial (i.e., sustentacular) cells; CGA and β3tub for neuroepithelial cells (Table 1, Fig. 1). Both the vascular and the neural PGL components were ultrastructurally aberrant (Fig. 1).

The vasculoneural paraganglioma network expresses mesenchymal, hypoxic, vasculoneurogenic and metabolic markers and is ultrastructurally aberrant. a ZEB1 is evident in the nuclei of both neuroepithelial (NE) and vascular cells (V), the latter identified by proximity to CD34-positive capillaries. b Ultrastructural relationship between an atypical capillary, with prominent endothelial cells (EC), and a cohesive nest of neuroepithelial cells (NE) with vacuolated cytoplasm, swollen mitochondria, neurosecretory granules and notched, lightly clumped nuclei (N). c–d Vascular distribution of β2-microglobulin (B2M), nestin and VEGFR1. f The hyaluronan receptor CD44 outlines the membranes of sustentacular (SC) and neuroepithelial (NE) zellballen cells. e Ultrastructural relationships between the four major cell types that organize the paraganglioma microenvironment: EC endothelial cells, PE pericytes, SC sustentacular cells, NE neuroepithelial cells. g Nuclear and cytoplasmic HIF2A is mainly evident in nestin-negative non-vascular cells. h Strong expression of the the insulin-regulated glucose transporter GLUT4 is mainly evident in neuroepithelial cells (NE). i Ultrastructural context showing neuroepithelial nests (NE) and adjacent capillaries with endothelial cells (EC) and pericytes (PE). DAPI (blue) identifies nuclei in the ApoTome immunofluorescence panels, dashed lines link immunofluorescence images to comparable ultrastructural areas. Bars = 10 µm (ApoTome immunofluorescence) and 5 µm (transmission electron microscopy). N nuclei
Ex vivo flow cytometry further substantiated the presence of consistent PGL cell populations positive for mesenchymal and stem cell markers (CD44, CD73, CD90, CD105, CD133) [14, 74, 75] (Online Resource 1, Tables S4, S5; Online Resource 2, Fig. S2) and demonstrated that variable subsets of PGL cells expressed hematopoietic/endothelial (CD34), glial (GFAP) and/or neural (NCAM) stem/progenitor cell markers. However, in most of the examined cases, the cells positive for CD34 comprised subsets coexpressing stem cell (CD133/CD44), neural (NCAM) or glial (GFAP) markers, suggesting phenotypic transition potential (Online Resource 1, Tables S5, S6).
These findings, replicated in tumours from SDHx carriers and noncarriers (Table 1; Online Resource 1, Tables S1, S5, S6), indicate that PGLs, regardless of the SDHx genetic background of the patient, contain immature cells with hybrid mesenchymal–vasculoneural features, likely primed for vascular (endothelial/mural) or neural (glial/neuroepithelial) differentiation.
It has long been known that PGLs may harbour ultrastructural mitochondrial aberrations predictive of reduced OXPHOS [69]. We, therefore, investigated the fine mitochondrial ultrastructure of the various PGL cell types. This revealed that hyperplastic and swollen mitochondria with disrupted cristae selectively accumulated in the neuroepithelial cells, a characteristic found in all the 77 PGLs examined by TEM (Fig. 2a, b). Comparing the cross-sectional areas of the mitochondrial profiles, it was nonetheless evident that larger mitochondria were significantly associated with PGLs from carriers of mutations in SDHB/C/D and to, to a lesser extent, SDHA, while the mitochondria associated with SDHAF2 mutation carrier status (unique case) were actually smaller than those of the PGLs from noncarriers (Fig. 2c). We next assessed the mt ΔΨ of the PGL tissue components using the JC-1 assay. The red-to-green fluorescence ratios, which reflect the mt ΔΨ [52, 73], decreased in the vascular PGL component relative to the autologous adipose tissue control and collapsed in the neuroepithelial component (Fig. 2d, e). This occurred in all of the 11 PGLs tested, with non-significant differences between the seven tumors from SDHx carriers and the four tumors from non-carriers (Fig. 2e). Thus, the neuroepithelial PGL lineage was characterized by severe mitochondrial dysfunction, implying that OXPHOS inactivation depended on the differentiation and/or the microenvironment.

Morphofunctional alterations of mitochondria in the neuroepithelial component of paraganglioma. a The transmission electron micrograph on the left shows the interface between normal inner ear epithelium (IEE, bottom half of the microscopic field) and an underlying non-embolized tympanic area of a tympano-jugular paraganglioma (PTJ64, from an SDHC mutation carrier, top half). The central higher magnification views illustrate the ultrastructure of the mitochondria in IEE cells (bottom) and in neuroepithelial paraganglioma cells (top). Mitochondrial hyperplasia with swelling and consequent loss of cristae is clearly evident in paraganglioma cells, while the mitochondria of the adjacent inner ear epithelial cells are structurally normal, which rules out the possibility that the mitochondrial alterations might have been due to surgical/post-surgical manipulation. The transmission electron micrograph on the right, focused on another PTJ64 paraganglioma field, illustrates the morphological differences between the roundish, swollen mitochondria with disrupted cristae routinely found in neuroepithelial cells (NE) and the smaller, relatively well-preserved mitochondria found in the adjacent endothelial cells (EC). The capillary lumen contains an erythrocyte. Bars = 10 µm (left image) and 1 µm (centre and right images). b Scatter plots of mitochondrial cross-sectional areas (µm2) determined on electron micrographs of neuroepithelial (NE) and endothelial cells (EC) from 20 paragangliomas (69 measurements randomly extracted from a total of 354 measurements available for NE cells compared to 69 measurements for endothelial cells). c Scatter plots of mitochondrial cross-sectional areas (µm2) determined on electron micrographs of neuroepithelial (NE) paraganglioma cells presented according to germline SDHx status. Mean areas were: 0.258 ± 0.150 µm2 (noncarriers, NC, 168 measurements); 0.384 ± 0.318 µm2 (mutated in SDHA, 66 measurements, P < 0.05); 0.724 ± 0.573 µm2 (mutated in SDHB, 45 measurements, P < 0.01); 0.568 ± 0.465 µm2 (mutated in SDHC, 54 measurements, P < 0.001); 1.008 ± 0.480 µm2 (mutated in SDHD, 15 measurements, P < 0.001); 0.152 ± 0.087 µm2 (mutated in SDHAF2, 6 measurements, P > 0.05). P-values were calculated using one-way anova with Games-Howell post hoc test. d JC-1 fluorescence highlights collapse of the mitochondrial membrane potential in the neuroepithelial component. A set of 5 paragangliomas is shown, two from SDHx noncarriers (PTJ68, PTJ66) and three positive for germline mutation in SDHC (PTJ64 and PTJ58) and SDHB (PT69). Regions of high mt polarization (top panels, red to yellow fluorescence, indicative of concentration-dependent J-aggregates, arrows) largely correspond to the vascular/glial component, while the neuroepithelial nests and cords (NE) are depolarized, as indicated by the green fluorescence due to JC-1 monomers. Micrographs in the panels below show SDHB immunohistochemistry for the same tumors, where arrows point to the vascular/perivascular component that supports more or less defined neuroepithelial zellballens (ZB). Brown granular SDHB staining is undetectable in the neuroepithelial components of PT69, whereas PTJ58, PTJ64, PTJ66, PTJ68 are weakly to strongly positive. The variability in SDHB protein immunohistochemistry contrasts with the constant evidence of mt depolarization in the neuroepithelial component, which is unrelated to SDHx/SDHB status. Bars = 20 µm. Hematoxylin is used as a counterstain in SDHB immunohistochemistry. e The red/green JC-1 fluorescence ratios, indicative of mt membrane potential, decrease in the vascular (mean: 0.92 ± 0.04, all cases; 0.72 ± 0.06, carriers; 1.07 ± 0.05, noncarriers) and, more prominently, neuroepithelial (mean: 0.54 ± 0.02, all cases; 0.53 ± 0.06, carriers; 0.56 ± 0.02, noncarriers) paraganglioma components compared to the autologous white abdominal adipose tissue (WAT, mean: 1.25 ± 0.06, all cases; 1.18 ± 0.1, carriers; 1.28 ± 0.07, noncarriers). The differences between the neuroepithelial and the vascular tumour components and between the neuroepithelial tumour component and WAT are significant. Data are from 11 PGLs, of which 4 associated with identified germline SDHx mutations and 7 SDHx mutation-negative
Patient-derived paraganglioma cultures
We developed 18 primary and 4 immortalized cell cultures from 19 PGLs (Online Resource 1, Table S7). Compared to the dissociated PGL cells, these cultures demonstrated quite homogeneous flow cytometric profiles, characterized by diffuse and mostly strong positivity for the mesenchymal markers CD73, CD90, CD105, for the embryonic and neural stem cell markers SOX2 and nestin, and for GFAP and PDGFRA. CD34 and NCAM were less consistently expressed; CD133 was below detection level (Fig. 3a; Online Resource 1, Table S7). These characteristics were unaffected by immortalization and by the SDHx carrier status of the donor patient (Online Resource 1, Table S7).

Characteristics of the paraganglioma cell cultures. a Flow cytometry phenotype based on the expression of surface related (CD73, CD90, CD105, CD133, NCAM/CD56, CD34, PDGFRA) and intracellular (SOX2, Nestin, GFAP) antigens in a primary paraganglioma cell culture (PTJ114p, passage 6). Red and blue histograms show the distribution of antigen expression and background control, respectively. Data are representative of experiments performed on 18 primary and four immortalized paraganglioma cultures (see Online Resource 1, Table S7). b ApoTome immunofluorescence analysis of representative paraganglioma cell cultures PTJ64p and PTJ86p illustrates positivity for: HIF2A (cytoplasmic and nuclear); ZEB1 (nuclear); smooth muscle actin (SMA) versus vimentin; nestin (filamentous cytoplasmic) versus PDGFRA (punctate pattern: membrane, cytoplasm, nucleus); VEGFR2 (membrane, cytoplasm and nucleus) versus CD44 (membrane and cytoplasmic); VEGFR1 versus DLK1 (both with membrane, cytoplasmic and nuclear distribution); CD34 (membrane and cytoplasmic); β3-tubulin (filamentous cytoplasmic); NCAM/CD56 (membrane and cytoplasmic). DAPI is used as a nuclear marker, bars = 10 µm. c Tridimensional growth in a primary culture (PTJ64p). From left to right: tridimensional focus at subconfluence (phase contrast); optical ApoTome immunofluorescence section illustrating vimentin positivity in fusiform peripheral cells and nestin positivity in inner cells; higher magnification field showing residual vimentin in cytoplasmic vacuoles of nestin-positive cells, suggestive of lysosomal degradation. DAPI is used as a nuclear marker, bars = 30 µm (left) and 10 µm (center and right). d Vasculoangiogenic potential. From left to right: phase contrast microscopy shows that already at 4 h from seeding on Matrigel paraganglioma cultures self-organize into a web-like network suggestive of microvascular capillary cords; confocal microscopy illustrates positivity for CD34, PDGFRA and DLK1. Images are from PTJ84p (from an SDHx noncarrier), similar results (not shown) were obtained for PTJ62p, PTJ66p, PTJ67p (all from noncarriers) and PTJ86p (from an SDHD mutation carrier). Bars = 100 µm
By AIF the PGL cultures, both primary and immortalized, diffusely exhibited the immature mesenchymal, hypoxic and vasculoneural markers shared by the vascular and neural components of the PGLs of origin, CD34 was detectable in few cells, β3tub was heterogeneously expressed, CGA was undetectable (Fig. 3b; Table 1). In the tridimensional foci randomly formed in adhesion vimentin was expressed in cells of the outer shell, SMA and nestin in inner cells, not directly exposed to the culture medium (Fig. 3c). When plated on Matrigel [19] the cells rapidly generated pseudovascular networks expressing the putative endothelial marker CD34 together with DLK1 and PDGFRA, key components of the signalling mechanisms that engage mural cells to nascent endothelial tubes [11, 37, 55, 62, 64] (Fig. 3d). Under non-adhesive conditions the cultures formed neurospheres (Online Resource 2, Fig. S3a), that strongly expressed the stem/mesenchymal markers found in the original PGL biospecimens and in the adherent cultures (Table 1; Online Resource 2, Fig. S3b). Notably the cultured cells, both primary and immortalized, contained ultrastructurally normal tubular mitochondria with mainly red JC-1 fluorescence (high mt ΔΨ), implying functional OXPHOS (Online Resource 2, Fig. S4). Thus, the mitochondrial dysfunction noted in the neuroepithelial component of the PGLs was not conserved in the mesenchymal-like cells that developed under culture conditions.
High ZEB1 and PDGFRA levels in PGL cells are a consequence of microRNA dysregulation
Our previously reported evidence that the miR-200a,b,c and the miR-34b are consistently downregulated in PGLs [7] contributes to explain why PGLs and derived cell cultures express constitutively high levels of two proteins that are known to be targeted by these miRs, NOTCH1 and ZEB1 [7, 70]. We previously validated the NOTCH1 gene as a miR-200 and miR-34 target in PGL cells [7], but ZEB1 remained to be confirmed in the specific context. Considering additional targets, we noted that the PDGFRA 3′UTR (cDNA NM-006206) contains four predicted binding sites for the miR-141/200a (Fig. 4a) and that miR-34a and -34c reportedly downregulate PDGFRA in lung cancer cells [21]. Using SH-SY5Y neuroblastoma cells, transfectable with miR mimics, we demonstrated that PDGFRA protein expression decreased by about 30% with miR-34c-5p mimic and by 99.96 and 32% with miR-200a-3p and miR-200b-3p mimics, respectively. No reduction was observed with miR-200c-3p mimics (Fig. 4b). To test direct interactions with the PDGFRA 3′UTR, the predicted wild-type and mutant miR-200a-3p target sites of the 3′UTR-PDGFRA cDNA were cloned downstream of the psiCHECK2 luciferase reporter vector and co-transfected with miRNA mimics into SH-SY5Y cells. Significant reductions in the luciferase activity of the vector carrying the wild-type PDGFRA-3′UTR, confirmed at three of four sites (Fig. 4c), demonstrated that miR-200a-3p directly targets PDGFRA.

MiR-200 and miR-34 family members target PDGFRA and ZEB1 in paraganglioma cells and induce cell death. a Schematic structure of the PDGFRA cDNA with location of four putative binding sites for miR-141/200a and of two for miR-34ac/449 (Targetscan). b Western blot analysis of PDGFRA (upper panel) and β-actin (loading control, bottom panel) after the indicated microRNA mimic transfections in SH-SY5Y neuroblastoma cells. Normalization with densitometric analysis is shown. c Relative luciferase activity dependent on miR-200a mimic transfections in SH-SY5Y cells is also shown (WT wild type, MUT mutant). d Western blot analysis of PDGFRA, ZEB1 and loading controls (β-actin and vinculin) in PTJ64i cells infected with lentivirus transducing either miR-200s or miR-34s or scramble. Normalization with densitometric analysis is shown. e, f ApoTome immunofluorescence, which distinguishes lentivirus-infected (GFP-positive) from non-infected (GFP-negative) PTJ64i cells, shows loss of PDGFRA and BAX expression in GFP-positive cells infected with lentivirus transducing miR-34bc or miR-200ab/429 compared to scramble control
To validate ZEB1 and PDGFRA as miR-200 and/or miR-34 targets in a PGL cell context, we enforced miR-200 (LV-200ab/429) or miR-34 (LV-miR-34bc) overexpression by lentiviral infection in PTJ64i cells. As expected, miR-200ab/429 reduced both the PDGFRA and the ZEB1 protein levels (70 and 60% respectively), while miR-34bc strongly reduced the PDGFRA protein level (89%), but did not affect ZEB1 (Fig. 4d). AIF analysis corroborated these data, showing that the loss of PDGFRA signal following the reintroduction of the miRs was accompanied by an upregulation of BAX protein expression, which pointed to the activation of an apoptotic response (Fig. 4e, f).
PDXs arise via vasculo-angio-neurogenesis from mesenchymal-like cells
PDX formation is predicted to depend on cells that elude ischemic necrosis, a consequence of prolonged devascularization during heterotransplantation and engraftment [26, 44, 63]. To highlight such cells, we analysed ex vivo cultured PGL tissues, a model that mimics the microenvironment of the post-transplantation phase preceding xenograft vascularization. Confocal and semithin section light microscopy coupled with TEM showed that by day 10 post-surgery the tumour tissue underwent widespread coagulative necrosis with loss of all vascular and neural cells, but some areas were recolonized by vimentin-positive mesenchymal-like cells, characterized by pseudopodia and filopodia, indicative of motility and phagocytic capability (Online Resource 2, Fig. S5a). Such endogenous cells remodelled the necrotic scaffold through phagocytosis and deposition of collagenous matrix and formed CD34-positive pseudovascular structures. These were the only cells identifiable in the ex vivo-cultured PGL samples and were similar to those observed in an early phase of PDX development (3 weeks; Online Resource 2, Fig. S5b), which suggests that they correspond to the cells responsible for PDX initiation.
We next developed PDX models to investigate PGL development in vivo. A total of 90 PGL fragments from 16 patients were xenografted into immunodeficient mice, with a high overall take-rate of 89% (80/90), not affected by the SDHx mutation carrier status of the donor patient (Online Resource 1, Table S8).
At harvest, 4.5–10 months post-transplantation, the PDXs presented as ~ 4–6 mm nodules, which infiltrated the adjacent murine neurovascular bundles, as typical of PGLs [47, 53, 61] (Online Resource 2, Fig. S6a). The relatively small size was consistent with the well-known slow growth of PGLs in patients (0–2 mm/year) [47, 53, 61]. The PDX tissue, including the vasculature, was of human origin, as proved with human-specific antibodies and mt DNA analysis, and was linked to the systemic murine circulation, as demonstrated by its permeation with India ink solution after intracardiac perfusion (Online Resource 2, Fig. S6).
TEM and AIF revealed the fine morphological details of the vasculo-angiogenic processes taking place in the PDXs (Fig. 5). These initiated in individual endothelial precursors, that progressively developed intracytoplasmic lumina through vacuolization (i.e., cell hollowing), as reported in HUVECs and in drosophila and zebrafish embryos [16, 32, 35, 65, 72], mutually interacted and coalesced, forming lumenized tubules positive for β2-microglobulin, CD31 and CD34, as PGL endothelium (Fig. 5a; Table 1; Online Resource 2, Fig. S6c). The self-assembly of these tubular endothelial structures defined a novel perivascular niche, readily populated by as yet undifferentiated mesenchymal-like cells that tended to adhere to the abluminal endothelial membranes. Whole mount confocal microscopy revealed that the adhesion process was associated with dichotomic tubular branching, a remodelling pattern consistent with intussusceptive angiogenesis, a major form of nonsprouting angiogenesis that allows rapid bifurcation of neoformed vessels via endothelial invagination [45] (Fig. 5b). Endothelial intussusceptions dividing the original lumen into two newly formed minor lumens were confirmed by TEM (Fig. 5a, right panels). Of note, plasma membrane NOTCH1 was uniquely associated with the ablumenal aspect of the endothelium, in direct contact with closely adherent perivascular cells that expressed the HIF-induced non-canonical NOTCH antagonist DLK1, a cancer pericyte-related antigen, together with SMA and PDGFRA (Fig. 5c) [6, 11, 27, 39, 55, 62]. Such cells revealed ultrastructural features of mural cells (vascular smooth muscle cells or pericytes) (Fig. 5c).

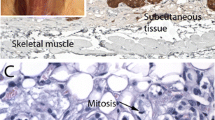
Patient-derived xenografts reveal endothelial tubulogenesis through cytoplasmic hollowing and vascular ramification via intussusceptive angiogenesis. a The transmission electron micrographs on the left show an early stage of lumenization, where endothelial precursors, with expanding cytoplasmic hollows (narrow arrows), associate (broad arrow) and coalesce in tubes (asterisk marks electron-dense, irrelevant intralumenal proteinaceous material). The semithin frozen section ApoTome images on the centre illustrate β2-microglobulin (B2M) positivity in a de novo formed endothelial tube versus control serial section (Cx). The transmission electron micrographs on the right illustrate intussusceptive pillar formation due to an endothelial infolding that splits the lumen into two newly formed lumens (red arrows indicate divided flow). Intraluminal India ink particles, visible in the higher magnification views on the far right (black arrows), indicate that the vessel undergoing remodelling is connected to the systemic murine vasculature. A dashed line links the tip of the intussuscepted endothelial cell to its context. Bars = 10 µm. b Mural cell recruitment and dichotomic vascular branching. Intravital whole mount JC-1 confocal microscopy illustrates lumenized endothelial tubes (asterisk: longitudinal or cross sections of vascular lumens) that attract assumably motile perivascular mesenchymal-like cells. Such cells, that have functional mitochondria, as indicated by JC-1 staining, are caught in the process of adhesion to the ablumenal side of the endothelial tubes (thin arrows). Recruitment of perivascular cells is associated with Y-shaped vascular branching (thick arrows). Bars = 10 µm (left images) and 20 µm (right image). c Characterization of the interaction between endothelial and mural cells during xenograft vasculogenesis. The transmission electron microscopy image on the right illustrates a vascular cross-section, with prominent mural cells, characterized by ovoid, scarcely clumped nuclei and clear, microfilament-rich cytoplasm (long arrow). Such cells wrap two smaller, darker endothelial cells that delimit a contracted lumen (short arrow). The pericyte-endothelial contact follows a jagged line (short arrow), where the endothelial and the mural plasma membranes are separated by a narrow gap. By ApoTome immunofluorescence, serial semithin sections of a similar vascular cross section are labelled with: CD31, which stains the endothelial cells only; smooth muscle actin (SMA), which stains the mural cells; NOTCH1 and SMA, which highlight the jagged abluminal endothelial membranes and the mural cells, respectively; VEGFR1, which stains both the mural cells and the endothelium; PDGFRA, which stains the mural cells and some adjacent interstitial cells; and, finally, DLK1 and NOTCH1. DLK1, a noncanonical NOTCH ligand, labels the mural cells in contact with the NOTCH1-positive ablumenal endothelial membranes. Homogeneous staining of intralumenal proteinaceous amorphous material (asterisk) is a fluorochrome-related artefact. DAPI is used as a nuclear marker in the ApoTome immunofluorescence images. Bars = 5 µm (transmission electron microscopy) and 10 µm (ApoTome immunofluorescence)

Ultrastructural and immunomorphological analogies between parangaglioma and derived xenograft. a ApoTome immunofluorescence of serial sections shows similar expression of vimentin, HIF2A, VEGFR2, DLK1, PDGFRA, ZEB1, VEGFR1 and β3-tubulin in paraganglioma (PGL) and patient-derived xenograft (PDX). Bars = 10 μm. DAPI is used as a nuclear marker. b Ultrastructural analogy between the neuroepithelial cell nest of a paraganglioma (PGL) and that of a corresponding patient-derived xenograft (PDX). The cohesive nests are structurally similar, except for the absence of neurosecretory granules in the PDX cells (Bars = 2 μm). c Similar mitochondrial alterations (swelling and loss of cristae) are visible in the neuropithelial cells of the paraganglioma (PGL) and of the derived xenograft (PDX) (Bars = 1.5 μm)
The early vasculo-angiogenic network developed on autonomously synthesized extracellular matrix (ECM) that initially contained collagen I, an EMT-related adhesive collagen associated with invasive growth and neovascularization [3, 51]. Then, with the recruitment of mural cells, collagen IV, main component of basement membranes [49, 60, 76], was deposited at the endothelial–mural interface and in the perivascular region (Online Resource 2, Fig. S7). This microenvironmental change was connected with the formation of clusters or ribbons of polygonal cells bound by glia-like spindle cells, that derived from the interstitial cells not involved in vascular differentiation. The de novo-formed cell nests were strongly reminiscent of the neuropithelial PGL component and exhibited the same immature mesenchymal, hypoxic and vasculoneurogenic markers found in neuropithelial PGL cells, together with β3Tub, despite negativity for the advanced neuroendocrine markers CGA and SYP (Fig. 6a, Table 1). Notably, the neuroepithelial-like cells of the PDX harboured hyperplastic and swollen mitochondria with disrupted cristae, indicative of context-dependent mitochondrial dysfunction, as in the neuroepithelial clusters of the PGLs (Fig. 6b, c).
Imatinib blocks PGL cell growth and inhibits xenograft formation
A low dose of imatinib (10 μM) significantly inhibited the growth of 4 PGL cell cultures (PTJ84p, PTJ86p, PTJ64p and the immortalized derivative PTJ64i), selected to represent PGLs from patients with diverse SDHx carrier status (Fig. 7a; cultures listed in Online Resource 1, Table S7). In PTJ64i cells, that could be grown in large numbers, downregulation of the ZEB1, PDGFRA and PDGFRB proteins, global protein dephosphorylation and an upregulation of Beclin 1 were evident at 48 h using ApoTome immunofluorescence and western blot (Fig. 7b, c). Activation of apoptosis was supported by evidence of BAX protein expression detected via ApoTome immunofluorescence at 48 h of imatinib treatment (Fig. 7b) and by a caspase assay at 15 h (Fig. 7c). Imatinib also induced significant mainly upward variation in the expression of miR-200a/b/c and miR-34b/c (Online Resource 2, Fig. S8), that may contribute to explain the downregulation of the ZEB1 and PDGFRA proteins.

Imatinib arrests paraganglioma cell growth and prevents paraganglioma xenograft formation. a Imatinib at 10 μM concentration arrests the growth of paraganglioma cultures. Cultures include PTJ84p, from an SDHx noncarrier (NC); PTJ86p, mutated in SDHD; PTJ64p, mutated in SDHC, and PTJ64i, immortalized with SV40-hTERT. Results are means ± standard deviations of 3 replicates. b ApoTome immunofluorescence analysis. At 48 h of treatment with 10 μM imatinib versus vehicle PTJ64i cells show loss of ZEB1 and PDGFRA, while BAX becomes evident (DAPI is used as a nuclear marker, bars = 10 µm). c Western blot and caspase activity. Imatinib (10 μM) inhibits global protein tyrosine phosphorylation, downregulates ZEB1 and upregulates Beclin-1 in PTJ64i cells at 48 h of treatment (normalization relative to β-actin calculated with NIH ImageJ). In the same cells caspase 3/7 activity increases at 15 h. d Morphology of the PTJ64i cell xenografts formed in immunodeficient mice treated with imatinib versus vehicle. Tumour xenografts were identified at 2/10 PTJ64i transplant sites in the imatinib-treated group (50 mg/Kg for 20 days, then 16.6 mg/Kg for 20 additional days) versus 11/12 PTJ64i transplant sites in the vehicle-trated group (P = 0.0015). In the vehicle-treated group (upper panel images), the xenografts presented as brownish plaques within 5 mm of diameter. Transmission electron microscopy highlighted morphological features similar to those of the patient-derived tumour xenografts, with blood vessels lined by prominent endothelial cells (EC) associated with zellballen-like neuroepithelial nests delimited by glia-like fusiform cells. The far right image shows a well-formed blood vessel with endothelial (EC) and mural (MC) cells (N nuclei). The only two xenografts detected in the imatinib-treated group (exemplified in the lower panel images) were smaller in size (within 3 mm) and, by transmission electron microscopy, contained only poorly organized cells with severe cytoplasmic vacuolization and accumulation of autophagosomes (Au), suggesting suppressed autophagic turnover (N nuclei). There was no evidence of vascular and neuroepithelial structures
We then verified whether early imatinib treatment, starting at day 3 from heterotransplantation, could inhibit in vivo xenograft formation. To this end PTJ64i cells were injected into the flanks of CD1 mice (11 mice, 22 transplants), which were then randomly assigned to imatinib-treated (5 mice, 10 transplants) and vehicle-treated (6 mice, 12 transplants) groups. At harvest, 7 weeks post-transplantation, CDXs were detected at 11/12 control transplant sites. The CDXs consisted in reddish-brown, flat patches of 4–6 mm in diameter that, as the above-described PDXs, comprised a vasculo-angiogenic network supporting nests of neuroepithelial-like cells (Fig. 7d, upper panels). As in the original PGLs, these cells harboured hyperplastic and swollen mitochondria, not present in the endothelial cells (Online Resource 2, Fig. S4b). In contrast, only 2 CDXs were identified at 10 heterotransplant sites of the imatinib-treated mice (P = 0.0015 by two-tailed Fisher exact test). These contained only disorganized autophagic or apoptotic cells, with no signs of an ongoing vasculogenic process (Fig. 7d, lower panels). The in vivo evidence of severe autophagy was consistent with the upregulation of Beclin 1, previously observed in the in vitro treated PTJ64i cells. Standard macroscopic and microscopic analysis of the thoracoabdominal organs did not reveal signs of imatinib toxicity (data not shown). A trend of body weight decrease (average 10%) was noted in the treated group of mice during the first 20 days of treatment, when the mice were injected with imatinib at 50 mg/Kg. Weight was regained in the following 20 days, when the imatinib dose was lowered to one-third (16.6 mg/Kg).
Discussion
We show here that PGLs are biphasic vascular and neural tumors sustained by stem-like mesenchymal cells primed for bipotent vascular and neural differentiation. These tumors appear to originate through an endogenous developmental program, schematically recapitulated in Fig. S9 (Online Resource 2), where the spatiotemporally coordinated emergence of the two interdependent tissue compartments, both of neoplastic origin, leads to the organization of the neoplastic microenvironment and to predictable mitochondrial changes. This program appears to be independent of the SDHx carrier status of the patients, and impacts on preventive and therapeutic interventions, which should be primarily directed against vasculoangiogenesis [22, 28], an early and fundamental phase of PGL tissue organization.
Our conclusions are based on convergent data from SDHx-characterized ex vivo, in vitro and in vivo models, including original PGLs, derived cell cultures, cell- and patient-derived xenografts (CDXs and PDXs). These integrative models shed light on the nature of the PGL-initiating cells and on the initial phases of PGL growth.
The PGL histogenetic sequence contrasts with the concept that PGL is primarily a neural tumour [71] and, in general, with the notion that tumour-derived vasculogenesis (vasculogenic mimicry) occurs late in the neoplastic progression [8, 23, 40]. However, it has been shown that primary neoplastic vasculogenesis characterizes the hemangioblastomas associated with von Hippel-Lindau disease, a syndrome that may include PGL [79]. Furthermore, tumour-derived blood vessels were recently and independently reported in PDXs obtained from trunk PGLs and pheochromocytomas [53]. On the other hand, the interconvertibility of mesenchymal stem cells with glial cells has been demonstrated in experimental models [36].
Vasculo-angiogenesis is negatively controlled by miRNAs belonging to the miR-200 and miR-34 families, that antagonize the EMT and promote neural differentiation [25, 48, 50, 70, 77]. We previouly reported that the miR-200a,b,c and -34b,c, decreased in PGLs relative to parasympathetic nerve, control the proliferation and survival of PGL cells [7]. Here we show that these miRs directly and/or indirectly control ZEB1 and PDGFRA in the context of PGL cells, as we previously demonstrated for NOTCH1 [7]. NOTCH1, ZEB1 and PDGFRA are key mediators of vasculo-angiogenesis and radio/chemoresistance [1, 34, 58, 68, 70, 77, 78]. This implicates miRNA dysregulation in PGL biology and aggressiveness.
Regardless of germline SDHx mutation status, neuroepithelial differentiation within the PGLs and the PDXs/CDXs was associated with severe mitochondrial dysfunction, implying loss of OXPHOS activity and glycolytic dependence [52, 69, 73]. The mitochondrial alterations were not present in the in vitro cultured PGL stem-like mesenchymal cells, but were acquired in vivo, during PDX/CDX formation in the neuroepithelial cell differentiation context. In this regard it is known that transformed mesenchymal cells, due to their motility and energetic requirements, mainly rely on oxidative phosphorylation [20], whereas tight clusters of neuroepithelial cells, obviously immotile and vulnerable to hypoxic stress due to crowding, must decrease their dependence on OXPHOS, which is compensated by increasing glycolysis [12, 29].
In conclusion, the stem-like mesenchymal component of PGL self-determines its microenvironment, its differentiation and its metabolism according to an endogenous developmental program that may reflect that responsible for physiological paragangliar organogenesis [15, 16]. The biological aggressiveness of the tumour conforms to the inherent tissue regenerative capacities of the multipotent stem-like PGL cells, activated by ischemia and enhanced by resilient embryonic vasculo-angiogenic mechanisms. All this should confer resistance to radio/chemotherapy and standard antiangiogenic therapy [2, 5, 54, 66], and could account for PGL recurrence after treatments that induce ischemic necrosis, such as embolization [61]. However, the fundamental dependence of PGLs on a vasculo-angiogenic phase may provide a targetable Achilles’ heel [17, 43].
Change history
01 February 2018
The given and family names of two co-authors were incorrect in the published article. The correct spelling should read as: Sampath Chandra Prasad and Vinagolu K Rajasekhar.
References
Ahn YH, Gibbons DL, Chakravarti D, Creighton CJ, Rizvi ZH, Adams HP et al (2012) ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J Clin Investig 122:3170–3183. https://doi.org/10.1172/JCI63608
Aliabadi H, Vredenburgh JJ, Everson RG, Desjardins A, Friedman HS, McLendon RE et al (2010) Bevacizumab fails to treat temporal paraganglioma: discussion and case illustration. J Neurooncol 98:427–430. https://doi.org/10.1007/s11060-009-0091-2
Allen P, Melero-Martin J, Bischoff J (2011) Type I collagen, fibrin and PuraMatrix matrices provide permissive environments for human endothelial and mesenchymal progenitor cells to form neovascular networks. J Tissue Eng Regen Med 5:e74–e86. https://doi.org/10.1002/term.389
Bacciu A, Medina M, Ait Mimoune H, D’Orazio F, Pasanisi E, Peretti G et al (2015) Lower cranial nerves function after surgical treatment of Fisch Class C and D tympanojugular paragangliomas. Eur Arch Otorhinolaryngol 272:311–319. https://doi.org/10.1007/s00405-013-2862-9
Barker HE, Paget JT, Khan AA, Harrington KJ (2015) The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer 15:409–425. https://doi.org/10.1038/nrc3958
Boyle SC, Liu Z, Kopan R (2014) Notch signaling is required for the formation of mesangial cells from a stromal mesenchyme precursor during kidney development. Development 141:346–354. https://doi.org/10.1242/dev.100271
Cama A, Verginelli F, Lotti LV, Napolitano F, Morgano A, D’Orazio A et al (2013) Integrative genetic, epigenetic and pathological analysis of paraganglioma reveals complex dysregulation of NOTCH signaling. Acta Neuropathol 126:575–594. https://doi.org/10.1007/s00401-013-1165-y
Cao Z, Bao M, Miele L, Sarkar FH, Wang Z, Zhou Q (2013) Tumour vasculogenic mimicry is associated with poor prognosis of human cancer patients: a systemic review and meta-analysis. Eur J Cancer 49:3914–3923. https://doi.org/10.1016/j.ejca.2013.07.148
Cassol CA, Winer D, Liu W, Guo M, Ezzat S, Asa SL (2014) Tyrosine kinase receptors as molecular targets in pheochromocytomas and paragangliomas. Mod Pathol 27:1050–1062. https://doi.org/10.1038/modpathol.2013.233
Chan KC, Ko JM, Lung HL, Sedlacek R, Zhang ZF, Luo DZ et al (2011) Catalytic activity of Matrix metalloproteinase-19 is essential for tumor suppressor and anti-angiogenic activities in nasopharyngeal carcinoma. Int J Cancer 129:1826–1837. https://doi.org/10.1002/ijc.25855
Chi Sabins N, Taylor JL, Fabian KP, Appleman LJ, Maranchie JK, Stolz DB et al (2013) DLK1: a novel target for immunotherapeutic remodeling of the tumor blood vasculature. Mol Ther 21:1958–1968. https://doi.org/10.1038/mt.2013.133
Chiche J, Rouleau M, Gounon P, Brahimi-Horn MC, Pouyssegur J, Mazure NM (2010) Hypoxic enlarged mitochondria protect cancer cells from apoptotic stimuli. J Cell Physiol 222:648–657. https://doi.org/10.1002/jcp.21984
Decaudin D, de Cremoux P, Sastre X, Judde JG, Nemati F, Tran-Perennou C et al (2005) In vivo efficacy of STI571 in xenografted human small cell lung cancer alone or combined with chemotherapy. Int J Cancer 113:849–856. https://doi.org/10.1002/ijc.20652
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D et al (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 8:315–317. https://doi.org/10.1080/14653240600855905
Egeblad M, Nakasone ES, Werb Z (2010) Tumors as organs: complex tissues that interface with the entire organism. Dev Cell 18:884–901. https://doi.org/10.1016/j.devcel.2010.05.012
Ellertsdottir E, Lenard A, Blum Y, Krudewig A, Herwig L, Affolter M et al (2010) Vascular morphogenesis in the zebrafish embryo. Dev Biol 341:56–65. https://doi.org/10.1016/j.ydbio.2009.10.035
Erber R, Thurnher A, Katsen AD, Groth G, Kerger H, Hammes HP et al (2004) Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J 18:338–340. https://doi.org/10.1096/fj.03-0271fje
Erguven M, Bilir A, Yazihan N, Korkmaz S, Aktas E, Ovalioglu C et al (2012) Imatinib mesylate decreases the cytotoxic effect of roscovitine on human glioblastoma cells in vitro and the role of midkine. Oncol Lett 3:200–208. https://doi.org/10.3892/ol.2011.434
Francescone RA 3rd, Faibish M, Shao R (2011) A Matrigel-based tube formation assay to assess the vasculogenic activity of tumor cells. J Vis Exp. https://doi.org/10.3791/3040
Funes JM, Quintero M, Henderson S, Martinez D, Qureshi U, Westwood C et al (2007) Transformation of human mesenchymal stem cells increases their dependency on oxidative phosphorylation for energy production. Proc Natl Acad Sci USA 104:6223–6228. https://doi.org/10.1073/pnas.0700690104
Garofalo M, Jeon YJ, Nuovo GJ, Middleton J, Secchiero P, Joshi P et al (2013) MiR-34a/c-dependent PDGFR-alpha/beta downregulation inhibits tumorigenesis and enhances TRAIL-induced apoptosis in lung cancer. PLoS One 8:e67581. https://doi.org/10.1371/journal.pone.0067581
Geevarghese A, Herman IM (2014) Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies. Transl Res 163:296–306. https://doi.org/10.1016/j.trsl.2014.01.011
Gilbertson RJ, Rich JN (2007) Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7:733–736. https://doi.org/10.1038/nrc2246
Gill AJ, Benn DE, Chou A, Clarkson A, Muljono A, Meyer-Rochow GY et al (2010) Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol 41:805–814. https://doi.org/10.1016/j.humpath.2009.12.005
Gill JG, Langer EM, Lindsley RC, Cai M, Murphy TL, Murphy KM (2012) Snail promotes the cell-autonomous generation of Flk1(+) endothelial cells through the repression of the microRNA-200 family. Stem Cells Dev 21:167–176. https://doi.org/10.1089/scd.2011.0194
Gu W, Hong X, Potter C, Qu A, Xu Q (2017) Mesenchymal stem cells and vascular regeneration. Microcirculation. https://doi.org/10.1111/micc.12324
Guichet PO, Guelfi S, Teigell M, Hoppe L, Bakalara N, Bauchet L et al (2015) Notch1 stimulation induces a vascularization switch with pericyte-like cell differentiation of glioblastoma stem cells. Stem Cells 33:21–34. https://doi.org/10.1002/stem.1767
Heldin CH (2013) Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal 11:97. https://doi.org/10.1186/1478-811X-11-97
Her YF, Maher LJ 3rd (2015) Succinate dehydrogenase loss in familial paraganglioma: biochemistry, genetics, and epigenetics. Int J Endocrinol 2015:296167. https://doi.org/10.1155/2015/296167
Hohenstein P, Pritchard-Jones K, Charlton J (2015) The yin and yang of kidney development and Wilms’ tumors. Genes Dev 29:467–482. https://doi.org/10.1101/gad.256396.114
Ii M, Nishimura H, Sekiguchi H, Kamei N, Yokoyama A, Horii M et al (2009) Concurrent vasculogenesis and neurogenesis from adult neural stem cells. Circ Res 105:860–868. https://doi.org/10.1161/CIRCRESAHA.109.199299
Iruela-Arispe ML, Beitel GJ (2013) Tubulogenesis. Development 140:2851–2855. https://doi.org/10.1242/dev.070680
Josson S, Nomura T, Lin JT, Huang WC, Wu D, Zhau HE et al (2011) Beta2-microglobulin induces epithelial to mesenchymal transition and confers cancer lethality and bone metastasis in human cancer cells. Cancer Res 71:2600–2610. https://doi.org/10.1158/0008-5472.CAN-10-3382
Kahlert UD, Suwala AK, Raabe EH, Siebzehnrubl FA, Suarez MJ, Orr BA et al (2015) ZEB1 promotes invasion in human fetal neural stem cells and hypoxic glioma neurospheres. Brain Pathol 25:724–732. https://doi.org/10.1111/bpa.12240
Kamei M, Saunders WB, Bayless KJ, Dye L, Davis GE, Weinstein BM (2006) Endothelial tubes assemble from intracellular vacuoles in vivo. Nature 442:453–456. https://doi.org/10.1038/nature04923
Kaukua N, Shahidi MK, Konstantinidou C, Dyachuk V, Kaucka M, Furlan A et al (2014) Glial origin of mesenchymal stem cells in a tooth model system. Nature 513:551–554. https://doi.org/10.1038/nature13536
Kim Y, Lin Q, Zelterman D, Yun Z (2009) Hypoxia-regulated delta-like 1 homologue enhances cancer cell stemness and tumorigenicity. Cancer Res 69:9271–9280. https://doi.org/10.1158/0008-5472.CAN-09-1605
Klein D, Meissner N, Kleff V, Jastrow H, Yamaguchi M, Ergun S et al (2014) Nestin(+) tissue-resident multipotent stem cells contribute to tumor progression by differentiating into pericytes and smooth muscle cells resulting in blood vessel remodeling. Front Oncol 4:169. https://doi.org/10.3389/fonc.2014.00169
Kofler NM, Shawber CJ, Kangsamaksin T, Reed HO, Galatioto J, Kitajewski J (2011) Notch signaling in developmental and tumor angiogenesis. Genes Cancer 2:1106–1116. https://doi.org/10.1177/1947601911423030
Krishna Priya S, Nagare RP, Sneha VS, Sidhanth C, Bindhya S, Manasa P et al (2016) Tumour angiogenesis-origin of blood vessels. Int J Cancer 139:729–735. https://doi.org/10.1002/ijc.30067
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15:178–196. https://doi.org/10.1038/nrm3758
Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C et al (2013) SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23:739–752. https://doi.org/10.1016/j.ccr.2013.04.018
Martin BJ (2013) Inhibiting vasculogenesis after radiation: a new paradigm to improve local control by radiotherapy. Semin Radiat Oncol 23:281–287. https://doi.org/10.1016/j.semradonc.2013.05.002
Maxson S, Lopez EA, Yoo D, Danilkovitch-Miagkova A, Leroux MA (2012) Concise review: role of mesenchymal stem cells in wound repair. Stem Cells Transl Med 1:142–149. https://doi.org/10.5966/sctm.2011-0018
Mentzer SJ, Konerding MA (2014) Intussusceptive angiogenesis: expansion and remodeling of microvascular networks. Angiogenesis 17:499–509. https://doi.org/10.1007/s10456-014-9428-3
Oudijk L, Neuhofer CM, Lichtenauer UD, Papathomas TG, Korpershoek E, Stoop H et al (2015) Immunohistochemical expression of stem cell markers in pheochromocytomas/paragangliomas is associated with SDHx mutations. Eur J Endocrinol 173:43–52. https://doi.org/10.1530/EJE-14-1164
Pacak K, Wimalawansa SJ (2015) Pheochromocytoma and paraganglioma. Endocr Pract 21:406–412. https://doi.org/10.4158/EP14481.RA
Pandey A, Singh P, Jauhari A, Singh T, Khan F, Pant AB et al (2015) Critical role of the miR-200 family in regulating differentiation and proliferation of neurons. J Neurochem 133:640–652. https://doi.org/10.1111/jnc.13089
Payne LS, Huang PH (2013) The pathobiology of collagens in glioma. Mol Cancer Res 11:1129–1140. https://doi.org/10.1158/1541-7786.MCR-13-0236
Pecot CV, Rupaimoole R, Yang D, Akbani R, Ivan C, Lu C et al (2013) Tumour angiogenesis regulation by the miR-200 family. Nat Commun 4:2427. https://doi.org/10.1038/ncomms3427
Peng DH, Ungewiss C, Tong P, Byers LA, Wang J, Canales JR et al (2017) ZEB1 induces LOXL2-mediated collagen stabilization and deposition in the extracellular matrix to drive lung cancer invasion and metastasis. Oncogene 36:1925–1938. https://doi.org/10.1038/onc.2016.358
Perelman A, Wachtel C, Cohen M, Haupt S, Shapiro H, Tzur A (2012) JC-1: alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis 3:e430. https://doi.org/10.1038/cddis.2012.171
Powers JF, Pacak K, Tischler AS (2017) Pathology of human pheochromocytoma and paraganglioma xenografts in NSG mice. Endocr Pathol 28:2–6. https://doi.org/10.1007/s12022-016-9452-5
Prasad SC, Mimoune HA, D’Orazio F, Medina M, Bacciu A, Mariani-Costantini R et al (2014) The role of wait-and-scan and the efficacy of radiotherapy in the treatment of temporal bone paragangliomas. Otol Neurotol 35:922–931. https://doi.org/10.1097/MAO.0000000000000386
Rafii S, Butler JM, Ding BS (2016) Angiocrine functions of organ-specific endothelial cells. Nature 529:316–325. https://doi.org/10.1038/nature17040
Rigoli L, Di Bella C, Verginelli F, Falchetti M, Bersiga A, Rocco A et al (2008) Histological heterogeneity and somatic mtDNA mutations in gastric intraepithelial neoplasia. Mod Pathol 21:733–741. https://doi.org/10.1038/modpathol.2008.58
Schermelleh L, Heintzmann R, Leonhardt H (2010) A guide to super-resolution fluorescence microscopy. J Cell Biol 190:165–175. https://doi.org/10.1083/jcb.201002018
Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR et al (2013) The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med 5:1196–1212. https://doi.org/10.1002/emmm.201302827
Soga T (2013) Cancer metabolism: key players in metabolic reprogramming. Cancer Sci 104:275–281. https://doi.org/10.1111/cas.12085
Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE (2009) Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood 114:5091–5101. https://doi.org/10.1182/blood-2009-05-222364
Taieb D, Kaliski A, Boedeker CC, Martucci V, Fojo T, Adler JR Jr et al (2014) Current approaches and recent developments in the management of head and neck paragangliomas. Endocr Rev 35:795–819. https://doi.org/10.1210/er.2014-1026
Tejada ML, Yu L, Dong J, Jung K, Meng G, Peale FV et al (2006) Tumor-driven paracrine platelet-derived growth factor receptor alpha signaling is a key determinant of stromal cell recruitment in a model of human lung carcinoma. Clin Cancer Res 12:2676–2688. https://doi.org/10.1158/1078-0432.CCR-05-1770
Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM et al (2012) Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol 9:338–350. https://doi.org/10.1038/nrclinonc.2012.61
Traustadottir GA, Jensen CH, Thomassen M, Beck HC, Mortensen SB, Laborda J et al (2016) Evidence of non-canonical NOTCH signaling: delta-like 1 homolog (DLK1) directly interacts with the NOTCH1 receptor in mammals. Cell Signal 28:246–254. https://doi.org/10.1016/j.cellsig.2016.01.003
Tung JJ, Tattersall IW, Kitajewski J (2012) Tips, stalks, tubes: notch-mediated cell fate determination and mechanisms of tubulogenesis during angiogenesis. Cold Spring Harb Perspect Med 2:a006601. https://doi.org/10.1101/cshperspect.a006601
van Beijnum JR, Nowak-Sliwinska P, Huijbers EJ, Thijssen VL, Griffioen AW (2015) The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol Rev 67:441–461. https://doi.org/10.1124/pr.114.010215
von Dobschuetz E, Leijon H, Schalin-Jantti C, Schiavi F, Brauckhoff M, Peczkowska M et al (2015) A registry-based study of thyroid paraganglioma: histological and genetic characteristics. Endocr Relat Cancer 22:191–204. https://doi.org/10.1530/ERC-14-0558
Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR et al (2010) Notch promotes radioresistance of glioma stem cells. Stem Cells 28:17–28. https://doi.org/10.1002/stem.261
Watanabe H, Burnstock G, Jarrott B, Louis WJ (1976) Mitochondrial abnormalities in human phaeochromocytoma. Cell Tissue Res 172:281–288
Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A et al (2009) The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol 11:1487–1495. https://doi.org/10.1038/ncb1998
Wick MR (2000) Neuroendocrine neoplasia. Current concepts. Am J Clin Pathol 113:331–335. https://doi.org/10.1309/ETJ3-QBUK-13QD-J8FP
Xu K, Cleaver O (2011) Tubulogenesis during blood vessel formation. Semin Cell Dev Biol 22:993–1004. https://doi.org/10.1016/j.semcdb.2011.05.001
Yamanaka R, Tabata S, Shindo Y, Hotta K, Suzuki K, Soga T et al (2016) Mitochondrial Mg(2+) homeostasis decides cellular energy metabolism and vulnerability to stress. Sci Rep 6:30027. https://doi.org/10.1038/srep30027
Yan Y, Zuo X, Wei D (2015) Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem Cells Transl Med 4:1033–1043. https://doi.org/10.5966/sctm.2015-0048
Yang Y, Guo L, Yang F, Huang Q, Zhang F, Ma H et al (2013) Implication of tumor stem-like cells in the tumorigenesis of sporadic paraganglioma. Med Oncol 30:659. https://doi.org/10.1007/s12032-013-0659-8
Yurchenco PD (2011) Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a004911
Zhang P, Sun Y, Ma L (2015) ZEB1: at the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 14:481–487. https://doi.org/10.1080/15384101.2015.1006048
Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J et al (2014) ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol 16:864–875. https://doi.org/10.1038/ncb3013
Zhuang Z, Frerich JM, Huntoon K, Yang C, Merrill MJ, Abdullaev Z et al (2014) Tumor derived vasculogenesis in von Hippel–Lindau disease-associated tumors. Sci Rep 4:4102. https://doi.org/10.1038/srep04102
Acknowledgements
This work was supported by the Italian Association for Cancer Research (AIRC), Grants IG9168 (2009–2012) and IG16932 (2015–2017) to RMC and by Ministry of Education, University and Research (PRIN) Grant #2015B7M39T_006 to AC. We gratefully acknowledge the services provided by the Mario Sanna Foundation Onlus, Piacenza, Italy, dedicated to the prevention and treatment of skull base tumors. We thank Professor Maria Addolorata Mariggio’, Unit of Functional Biotechnology, Center of Aging Science and Translational Medicine (CeSI-MeT), Gabriele d’Annunzio University, for kind help with mitochondrial studies; Ms. Rosalia De Amicis, Center of Aging Science and Translational Medicine (CeSI-MeT), Gabriele d’Annunzio University, for secretarial assistance; Ms. Anna Nassani, Department of Anatomic Pathology, Guglielmo da Saliceto Hospital, for expert help with immunohistochemistry.
Author information
Authors and Affiliations
Contributions
FV and SP developed paraganglioma cell culture and xenograft models; MS and SCP were the surgeons responsible for patient recruitment, clinical data and surgical sampling; SP, FV, MRP, CR, AS, RF, AM performed and/or analysed in vitro/in vivo experiments; SV, LT and LVL carried out immunomorphological and ultrastructural studies; DLE designed and performed western analyses; SG and SP designed and performed JC-1 assays; GB, MM, and PL carried out cytometric analyses; FS, ET and GO performed and analysed mutational studies; CTP was the pathologist who diagnosed the cases and performed and analysed immunohistochemistry; AV, RV, DLE and FV designed and analysed microRNAs’ experiments, performed by ADA, SP and AM; LVL, MS, CSN, AC and VKR analysed data and made intellectual contributions throughout. RM-C was the grant holder and principal investigator who supervised the studies and wrote the manuscript.
Corresponding author
Ethics declarations
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committees and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. In particular, the human biospecimens (tumour, blood, and, when possible, normal solid tissues), were collected in the operating room at surgery, with written informed consent following protocols approved by the G. d’Annunzio University ethics review board (protocol #841/10COET) and the Piacenza district health authority (A.USL Piacenza Ethical Committee, protocol #2016/6088). The consent forms included the request of permission to produce cell cultures and xenografts from tumour and normal tissue. For the studies with animals, all applicable international, national and/or institutional guidelines for the care and use of animals were followed. In particular, the in vivo experiments adhered to the regulations of the Italian law, the directives of the European Union and the Three R’s recommendations and were approved by the Italian Ministry of Health (protocols DGSAF-A-0022719 and F4738.2). The NSG mice were from a home-grown colony authorized by the Italian Ministry of Health (protocol DGSAF0024097).
Conflict of interest
The authors certify that they have NO affiliations with or involvement in any organization or entity with any financial or non-financial interest in the subject matter or materials discussed in this manuscript.
Additional information
The original version of this article was revised: The given and family names of two co-authors were incorrect in the published article as Sampath Chandra Prasat and Vinalogu K. Rajasekhar.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Verginelli, F., Perconti, S., Vespa, S. et al. Paragangliomas arise through an autonomous vasculo-angio-neurogenic program inhibited by imatinib. Acta Neuropathol 135, 779–798 (2018). https://doi.org/10.1007/s00401-017-1799-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1799-2