
Abstract
The most common cause of amyotrophic lateral sclerosis (ALS) is mutations in superoxide dismutase-1 (SOD1). Since there is evidence for the involvement of non-neuronal cells in ALS, we searched for signs of SOD1 abnormalities focusing on glia. Spinal cords from nine ALS patients carrying SOD1 mutations, 51 patients with sporadic or familial ALS who lacked such mutations, and 46 controls were examined by immunohistochemistry. A set of anti-peptide antibodies with specificity for misfolded SOD1 species was used. Misfolded SOD1 in the form of granular aggregates was regularly detected in the nuclei of ventral horn astrocytes, microglia, and oligodendrocytes in ALS patients carrying or lacking SOD1 mutations. There was negligible staining in neurodegenerative and non-neurological controls. Misfolded SOD1 appeared occasionally also in nuclei of motoneurons of ALS patients. The results suggest that misfolded SOD1 present in glial and motoneuron nuclei may generally be involved in ALS pathogenesis.
Similar content being viewed by others

Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease mainly characterized by progressive loss of upper and lower motoneurons and death usually ensues within 3–5 years after diagnosis. More than 150 mutations in the ubiquitously expressed enzyme superoxide dismutase-1 (SOD1) have been associated with familial ALS (FALS) and such mutations are found in about 6% of all ALS patients [35, 45] (http://alsod.iop.kcl.ac.uk). The mutations confer a cytotoxic property on the enzyme that is poorly understood [16]. Nine of the mutations cause long C-terminal truncations. These mutant SOD1s lack the stabilizing C57-C146 intrasubunit disulfide bond and also strand 8 in the β-barrel core of the protein, and thus cannot adopt native folding. The most extensively studied truncation mutant, Gly127insTGGG (G127X), has been found to be rapidly degraded and to be present only in minute amounts in the human spinal cord [22]. The mutant SOD1s associated with ALS should most likely cause ALS by essentially the same mechanism. This implies that any common cytotoxic conformational species of SOD1 should be misfolded and present in minute amounts in the tissue.
The wild-type human SOD1 can also have neurotoxic effects. Transgenic overexpression in mice leads to axonal abnormalities and a late moderate loss of ventral horn neurons [19, 24], and it exacerbates disease caused by mutant SOD1s [11, 19]. Compared to D90A, the most common of the ALS-associated mutant SOD1s, wild-type human SOD1 is between half and equally neurotoxic in mice [23]. Damage to this long-lived protein causes destabilization and results in interaction properties and toxicity similar to those found for mutant SOD1s [5, 34].
There is considerable evidence for the involvement of non-neuronal cells in the pathogenesis of ALS. In murine SOD1 models, down-regulation of SOD1 expression in astrocytes [47] and microglia [2] slows progression after disease onset, whereas down-regulation in motoneurons delays onset [4, 47]. In chimeric mice, motoneurons expressing mutant SOD1s are spared if surrounded by non-transgenic glial cells [10]. Neuron-restricted synthesis of G93A mutant SOD1 in mice is, however, sufficient to cause motoneuron pathology and a late ALS-like disease [20, 43]. Mutant SOD1 expression in oligodendrocytes might protect against loss of motoneurons [46]. The exact role of non-neuronal cells in the pathogenesis of human ALS is unknown, but all cells implicated in the transgenic models show signs of activation or alterations.
We have developed a set of antibodies which allow specific detection of misfolded SOD1 against the abundant background of native enzyme in the human CNS. Using these antibodies, we have previously shown that inclusions containing misfolded SOD1 are regularly present in motoneuron somas of ALS patients, both with and without SOD1 mutations [12]. The finding of misfolded SOD1 in sporadic ALS (SALS) was recently confirmed by Bosco et al. [5], and they also found that wild-type SOD1 immunopurified from SALS tissues inhibited fast axonal transport in a manner similar to H46R mSOD1 indicating a pathological mechanism common to SALS and SOD1 FALS. In this study, we searched for signs of SOD1 involvement in ALS focusing on non-neuronal cells. The major novel finding is that misfolded SOD1 is regularly present in the nuclei of ventral horn astrocytes, microglia, and oligodendrocytes in ALS patients carrying SOD1 mutations as well as in sporadic and familial patients lacking such mutations. Only negligible staining was seen in control patients with neurodegenerative and non-neurological disease.
Materials and methods
Subjects
Material was collected at autopsy from patients enrolled at the Department of Neurology, Umeå University Hospital. The group consisted of 43 patients with SALS [mean age 68 ± 12 (17–88) years] and 8 patients with FALS without SOD1 mutations [mean age 59 ± 11 (39–71) years]. The non-SOD1 FALS patients were subjected to genetic screening for mutations in the following genes; transactivation-responsive DNA-binding protein of molecular weight 43 kDa (TDP-43), fused in sarcoma (FUS), vesicle associated membrane protein B (VAPB), angiogenin and optineurin, and none were found. Moreover, nine ALS patients carrying SOD1 mutations were included in the study: six patients homozygous for the D90A mutation, two patients heterozygous for the G127X mutation, and one patient heterozygous for the D101G mutation [mean age 62 ± 11 (43–75) years]. ALS patients were diagnosed in accordance with the revised criteria of El Escorial [6]. In addition, two patients with spinobulbar muscular atrophy (SBMA) (aged 52 and 76 years, respectively) were included in the study.
Tissues were also collected from 26 control patients with other neurodegenerative diseases [mean age 74 ± 19 (2–92) years; 12 with Alzheimer’s disease; six with Parkinson’s disease; three with multiple sclerosis, one with tuberous sclerosis, one with Huntington’s disease, one with spinocerebellar ataxia 7, one with autosomal dominant progressive external ophthalmoplegia, and one with argyrophilic grain disease]. Tissues from 20 control patients without neurological disease [mean age 69 ± 17 (37–91) years; all patients died from heart conditions or pneumonia) were also examined. The post-mortem time for all patients was estimated to be between 0 and 3 days, and there was no difference in postmortem time between the groups. The study adhered to the tenets of the Helsinki Declaration, and was approved by the Ethical Committee of Umeå University. For detailed information on subject collection, see Electronic Supplementary Material.
Antibodies
For detection of misfolded SOD1, a set of polyclonal rabbit (Ra) antibodies (ab) raised against keyhole limpet hemocyanin-coupled peptides corresponding to amino acids 4–20 (Ra 4–20 ab), 57–72 (Ra 57–72 ab), and 131–153 (Ra 131–153 ab) in the human SOD1 sequence were used. A set of polyclonal chicken (Chi) antibodies corresponding to the same set of peptides was also used (Chi 4–20 ab, Chi 57–72 ab, Chi 131–153 ab, respectively). The antisera were affinity purified in two steps as described in previous papers [12, 23]. G127X mutant SOD1 has following Gly-127 a 5 amino acid long neopeptide before the C-terminal truncation. CNS material from G127X mutant SOD1-carriers was also stained with a mutant-specific Ra SOD1 peptide antibody directed against amino acids 123–132 in the mutant: CADDLGGQRWK, (neopeptide shown in bold). This antibody shows no reaction with wild-type human SOD1 [22]. Other commercially available antibodies used in the study are presented in Online Resource, Table S1.
Immunohistochemistry
After blocking, sections were incubated with primary antibodies and subsequently with corresponding fluorescent secondary antibodies or with biotin-conjugated secondary antibodies coupled to an avidin-horseradish peroxidase conjugate, and were visualized using aminoethylcarbazole as the precipitating enzyme product. The primary antibodies used were either the anti-SOD1 peptide antibodies described above or the antibodies shown in Online Resource Table S1. The sections were examined by confocal laser microscopy or using an Olympus BX50 light microscope. A four-tiered semi-quantitative scale was used to estimate the number of glial cell nuclei in each cross-section of spinal cord showing misfolded SOD1 staining. The levels were: 0 = no glial cells with nuclear staining; 1 = <25% of the glial cells showing nuclear staining; 2 = 25–75% of the glial cells showing nuclear staining; and 3 = > 75% of the glial cells showing nuclear staining. Sections from cervical, thoracic, and lumbar spinal cord were analysed for each patient and the result is presented in Table 1. Data for proportion of glial cell nuclei with staining were calculated as median (range). For detailed information on staining methods, see Electronic Supplementary Material.
Results
Antibody characterization
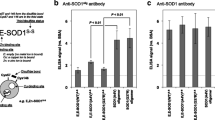
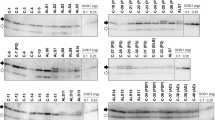
We have developed a set of rabbit and chicken antibodies against peptides in the human SOD1 sequence. The antibodies show high specificities for SOD1 in western immunoblots of human ventral horn extracts and they react only with misfolded SOD1 species in immunocapture experiments [12] (see also Online Resource, Fig. S1a, b). To further ascertain their specificities for misfolded SOD1 in immunohistochemical applications, the Ra 57–72 and the Ra 131–153 antibodies, and the Chi 57–72 and 131–153 antibodies, respectively, were subjected to preincubations. When pre-incubated with the appropriate immunizing peptide or with denatured SOD1, complete blocking of the signal was seen (Online Resource, Fig. S2b, d). Native SOD1 had no blocking effect (Online Resource, Fig. S2c). Exclusion of the primary antibodies resulted in the disappearance of all immunostaining. These studies show that the antibodies can be used for the specific detection of misfolded SOD1 species against the abundant background of natively folded SOD1 in tissues. In the following, positive immunohistochemical staining with these antibodies will be referred to as misSOD1 staining.
Misfolded SOD1 is found in the nuclei of glial cells in ALS patients carrying SOD1 mutations
Spinal cord sections from the nine ALS patients carrying SOD1 mutations were stained with the anti-SOD1 peptide antibodies. Aggregates/inclusions of misfolded SOD1 in the soma, axons, and dendrites of affected motoneurons were seen as previously described (Fig. 1d) [33, 36]. There was, however, also prominent staining of the nuclei of glial cells (Figs. 1a, d, 2a). The staining was composed of granules measuring 0.5–2 μm, which were dispersed throughout the nuclei. The immunohistochemical findings were identical using the different rabbit and chicken anti-SOD1 peptide antibodies, and the intranuclear SOD1 positive granules in glial cells were seen at all levels of the spinal cord and in both grey and adjacent white matter. The staining was detected with both bright-field and immunofluorescence microscopy (Figs 1a, 2a). Using a four-tiered semi-quantitative scale to estimate the proportion of glial nuclei with misSOD1 staining in one section, all patients homozygous for the D90A mutation showed misSOD1 staining in glial cell nuclei, ranging from less than 25% in two patients and up to almost 100% in four patients (Table 1). The D101G patient showed misSOD1 staining in approximately 25% of the glial cell nuclei (Table 1).

Misfolded superoxide dismutase-1 (misSOD1) is present in spinal cord ventral horn of ALS patients. Immunohistochemistry was performed with the Ra 131–153 ab in all cases. a Presence of misSOD1 in glial cell nuclei of a FALS patient homozygous for the D90A SOD1 mutation. b MisSOD1 in glial cell nuclei of a FALS patient not linked to SOD1 mutations. c MisSOD1 in glial cell nuclei of a SALS patient. A motoneuron also shows granular somal inclusions. d Higher magnification of a motoneuron from a patient homozygous for the D90A mutation, showing the presence of misSOD1 in the nucleus (arrow) and widely spread in the soma. Strongly stained glial nuclei are also seen. Motoneurons from a SALS patient showing intranuclear staining (e) and somal staining (f) of misSOD1. Strongly stained glial nuclei are also present in the section. g MisSOD1 in glial cell nuclei (arrows) and in motor neuron nuclei (arrowhead) from a patient with FALS not linked to SOD1 mutations. h Four of 46 control patients showed weak misSOD1-staining in spinal cord glial cell nuclei. As an example, the figure shows a non-neurological control patient. i Material from a control patient with Alzheimer’s disease showing no evidence of misSOD1. Scale bars are 50 μm (a, b), 30 μm (c, d), 20 μm (e, f) or 50 μm (g, h, i)

MisSOD1 staining is seen in human spinal cord ventral horn motoneurons and glial cells of ALS patients. All sections were stained with the Ra 57–72 ab. Presence of misSOD1 in glial cell nuclei (arrowheads) in a familial ALS (FALS) patient with the G127X SOD1 mutation (a), in a sporadic ALS (SALS) patient (b), and in a FALS patient not linked to SOD1 mutation (c). Higher magnification of different misSOD1 staining patterns in motoneurons from two SALS cases. The nuclear staining appears as granular aggregates approximately 0.5–3 μm in size, often seen in association with the nucleolus (d). In motoneurons with nuclear staining, the cytoplasm appears to contain less of the small misSOD1 inclusions. In panels b and e, misSOD1 aggregates can be seen scattered throughout the cytoplasm of motoneurons, and are particularly abundant in the somal area, while not penetrating the nuclear envelope (arrows). f MisSOD1 staining in a sample from a neurodegenerative control patient with Alzheimer’s disease. Glial cell nuclei and motoneurons lack misSOD1 aggregates/inclusions. Scale bars are 40 μm (a, b), 50 μm (c), 20 μm (d, e) or 30 μm (f)
Both patients with the G127X mutation showed nuclear staining of glial cells, but they differed in the proportion of cells that stained. Using the G127X-specific Ra 123–132 ab, misSOD1 staining was seen in <25% of the glial cell nuclei in both patients. One of the two patients also had some nuclear staining in motoneurons. Interestingly, the Chi 131–153 ab, raised against a peptide sequence that is absent in the G127X mutant SOD1, gave rise to misSOD1 staining in glial cell nuclei in both G127X patients, and in one of them it was seen in more than 75% of the glial cell nuclei. Double staining with the Chi 131–153 ab and the G127X mutant-specific ab mostly showed separate aggregates of misSOD1 staining, without co-localization (Online Resource, Fig. S3a–c). Performing double-staining with an antibody that detect both misfolded wildtype and G127X mutant SOD1 protein, the Chi 53–72 ab, with the mutant-specific G127X ab yielded sometimes co-localization as in large aggregates in motoneurons (Online Resource, Fig. S3d–f) but also single staining with the Chi 57–72 ab as in the arrow-head marked glial nucleus. This indicates that some of the misSOD1 staining within the glial cell nuclei of G127X patients has originated from wild-type SOD1.
To establish that the misSOD1 staining observed was present inside neuronal and glial nuclei, double immunofluorescence staining with an antibody to the structural nucleoporin protein NUP 62, which localizes to the nucleoplasmic region of the nuclear envelope, was performed [29]. Double staining with the NUP 62 antibody revealed the nuclear envelope, and inside it misSOD1 staining was seen (Online Resource, Fig. S4). MisSOD1 staining can thus be intranuclear in motoneurons and is principally intranuclear in glial cells.
Misfolded SOD1 is regularly found in the nuclei of glial cells and occasionally in motoneuron nuclei of ALS patients lacking SOD1 mutations
Sections of spinal cord from 43 patients with SALS and from 8 patients with FALS, all of whom lacked SOD1 mutations, as well as 2 patients with SBMA, were stained with the antibodies to SOD1 peptides. Intranuclear staining in glial cells was seen both in the SALS and the FALS patients. The staining patterns were virtually identical to those seen in the patients carrying SOD1 mutations. All sporadic ALS patients showed misSOD1 staining in their glial nuclei (Figs. 1c, e, 2b, 3a, d, g, 4a and Online Resource, Fig. S4f), as did seven of the eight FALS patients who lacked SOD1 mutations (Figs. 1b, 2c, 4d). We were unable to find nuclear misSOD1 staining in one FALS patient. On average, the proportion of glial cells in each section with misSOD1 staining was close to 75%, and ranged from <25% in some patients up to 100% in others (Table 1). The intranuclear SOD1 positive granules in glial cells were primarily seen in the ventral horn and adjacent white matter but some could also be observed in the dorsal horn and the dorso-medial white matter. Overall, the nuclear misSOD1 staining in glial cells was at least as prominent in patients lacking SOD1 mutations as in patients carrying such mutations (Table 1). Intranuclear staining for SOD1 in glial cells was also present in the two SBMA patients included in the study.

Aggregates of misfolded SOD1 are found in astrocytes, microglia and oligodendrocytes. Micrographs of lumbar ventral horn glial cells from two SALS patients (a–c and g–i) and a FALS patient homozygous for the D90A mutation (d–f) labeled with the Chi 131–153 ab (a, g) and the Ra 131–153 ab (d). (a–c) Double immunofluorescence labeling showing misSOD1 (green) and the astrocyte marker GFAP (red). In panel c, most of the glial cell nuclei containing misSOD1 are astrocytes. d–f Double immunofluorescence labeling showing misSOD1 (green) and the microglial marker Iba1 (red). In panel f, some of the glial cells with nuclear misSOD1 staining are microglia. g–i Double immunofluorescence labeling showing misSOD1 (green) and the oligodendroglial marker Olig 2 (red). In panel i, misSOD1 is present in the nuclei of oligodendrocytes. Scale bars are 20 μm (a–c) and 10 μm (d–i)

Aggregates of misfolded SOD1 in glial cell nuclei occasionally co-localize with ubiquitin, but not with TDP-43. Micrographs of cervical and lumbar spinal cord ventral horn sections from a SALS patient stained with the Chi 57–72 ab for misSOD1 (green) and with antibodies to ubiquitin and TDP-43 (red). MisSOD1 could be seen in glial cell nuclei (a) and in the cytoplasm of a motoneuron (d). Staining with the anti-ubiquitin antibody showed that some glial cell nuclei contained ubiquitin (b, arrows) and the merged picture (c) revealed some co-localization of ubiquitin and misSOD1 (arrows), although misSOD1 could also be seen separate from ubiquitin (arrowheads). Staining with anti-TDP-43 antibody revealed that some glial nuclei contained TDP-43 (e, arrows). Occasionally, misSOD1 and TDP-43 were present in the same glial cell nuclei (f) but no distinct co-localization of misSOD1 and TDP-43 was apparent. Scale bars are 20 μm (a–f)
Small and numerous aggregates of misfolded SOD1 were seen in the cytoplasm of motoneurons from ALS patients lacking mutations in the SOD1 gene, as we have previously reported [12]. These misfolded SOD1 aggregates were often scattered throughout the cytoplasm and were particularly abundant in the perinuclear area, but did not penetrate the nuclear envelope (Figs. 1f, 2b, e, 4d) [12]. However, occasionally motoneurons also showed nuclear misSOD1 staining. The staining then appeared as nuclear aggregates approximately 0.5–3 μm in size, often in association with the nucleolus (Figs. 1e, g, 2d and Online Resource, Fig. S4a). In motoneurons with nuclear staining, the cytoplasm often appeared to contain fewer aggregates of misfolded SOD1 (Fig. 1e). The motoneurons thus showed two types of misSOD1 staining: either intranuclear staining in motoneurons or a perinuclear misSOD1 staining with numerous aggregates of misfolded SOD1 in the soma but no sign of intranuclear staining (Fig. 2e). In the latter case, the nucleus appeared to be spared, leaving the impression that the nuclear envelope was acting as a barrier. The two types of motoneuron staining appeared to co-exist in the ventral horns and they could be seen in the same section, although somal and perinuclear staining was more common and was seen in all ALS patients investigated. On the other hand, the intranuclear staining in motoneurons was seen in 30 of 43 SALS patients and in six of eight FALS patients, all of whom lacked SOD1 mutations. Four out of the six patients homozygous for the D90A mutation and one of the two SBMA patients showed inclusions of misfolded SOD1 in the nuclei of motoneurons. The nuclear staining was only seen in a few motoneurons per section, and not in all sections investigated.
Nuclear staining of SOD1 in glial cells was specific for tissues affected by ALS and was not seen in controls with non-neurological or other neurodegenerative diseases
Spinal cord sections from 20 patients who died of non-neurological diseases were stained with the anti-SOD1 peptide antibodies. Only one patient showed misSOD1 staining in the nucleus of glial cells and motoneurons (Fig. 1h). The proportion of staining was <25% in that patient, and staining was not seen in all sections investigated (Table 1). Twenty-six controls with other neurodegenerative diseases were also examined. Twenty-three of these controls did not stain for misfolded SOD1 in ventral horn glial cell nuclei; nor was misfolded SOD1 detected in the nuclei of motoneurons (Figs. 1i, 2f). As the spinal cord is not the primary site of pathology in other neurodegenerative diseases, we also examined the brain areas mainly affected in Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease. We could not detect any misfolded SOD1 immunoreactivity in areas of the hippocampus, temporal, and frontal cortex, putamen, striatum, and substantia nigra (data not shown). The 4 of 46 controls that stained positive for misSOD1 showed increased GFAP staining in the spinal cord. All four also showed cytoplasmic inclusions in motoneurons when stained with ubiquitin, and in one neurodegenerative control, a patient with Alzheimer’s disease, a few skein inclusions could be seen in the motoneuronal cytoplasm as well.
Misfolded SOD1 is also found motoneurons and glial cells in brainstem and motor cortex
Mid-olivary sections from the brainstem from 12 neurodegenerative control patients, from 11 SALS patients, 2 non-SOD1 FALS patients and 8 SOD1 mutated patients were investigated for misfolded SOD1 by immunohistochemistry. In brain stem motor nuclei, we found intranuclear misSOD1 staining in glial cells in all ALS patients, although the number varied between patients. Staining was generally found in fewer glial cells than seen in the spinal cord. The staining was mainly found in motor nuclei and in the inferior olivary nucleus. We also found a primarily cytoplasmic staining in brain stem motoneurons in all ALS patients, while nuclear staining was only occasionally observed. We found no staining in the control patients investigated.
Sections from motor cortex were investigated in eight SALS patients. All patients had misSOD1 staining in glial cell nuclei and some cytoplasmic staining in neurons. The staining was less than found in brainstem and spinal cord.
Identification of glial cell types with nuclear staining of misfolded SOD1
Next, we wanted to identify the glial cell types carrying nuclei that were positive for misfolded SOD1. We used double-labeling immunofluorescence with the anti-SOD1 peptide antibodies and antibody markers for astrocytes, microglia, and oligodendrocytes. Using confocal laser scanning microscopy, we could show that most of the nuclei carrying misSOD1 staining were present in cells positive for the astrocytic marker GFAP (Fig. 3a–c). Double-labeled astrocytes were seen both in white and grey matter, and they were seen in all cases where positive staining for misfolded SOD1 was seen. We also found that some of the nuclei that were positive for misfolded SOD1 were present in cells that expressed the microglial marker Iba1 (Fig. 3d–f), although not all microglia had misfolded SOD1 in the nucleus. In addition, a few of the glial cell nuclei that stained for misfolded SOD1 expressed the olig 2 marker (Fig. 3g–i). These double-labeled oligodendroglia nuclei were much smaller and occurred less frequently than the astrocyte nuclei in the sections investigated.
SOD1-positive inclusions in glial cell nuclei occasionally co-localized with ubiquitin, but not with TDP-43 or p62
In the samples from SALS and FALS patients lacking SOD1 mutations stained with an antibody directed against TDP-43, cytoplasmic inclusions in motoneurons were seen in all sections investigated, as skein-like and/or fine granular punctuate inclusions, in accordance with the previous reports [1, 31]. For a more detailed description, see Electronic Supplementary Material. When ALS patients lacking SOD1 mutations were double-stained with the anti-SOD1 peptide antibodies, TDP-43 was not found to co-localize with the misfolded SOD1 aggregates seen in the somas of motoneurons. Occasionally, misfolded SOD1 and TDP-43 were located in the same glial or motoneuron nucleus but no co-localization could be demonstrated (Fig. 4d–f). Interestingly, two control patients, one with Alzheimer’s disease and another with cardiovascular disease, had TDP-43 positive neuronal cytoplasmic inclusions in the shape of skein inclusions in the cytoplasm of spinal cord motoneurons (data not shown). The staining was not different from that seen in SALS patients. Both of these controls were negative when staining for misfolded SOD1. The two SBMA patients also showed cytosolic TDP-43 staining typical of SALS patients (data not shown).
Since misfolded proteins present in the nuclei are degraded through the ubiquitin–proteasome pathway, we performed double immunofluorescence staining for misfolded SOD1 and ubiquitin. Occasionally, misfolded SOD1 co-localized with ubiquitin in glial cell nuclei, although most misSOD1 staining in glial cell nuclei was ubiquitin-negative (Fig. 4a–c).
The polyubiquitin-binding protein p62 (sequestosome 1) has been proposed to act as a factor for shuttling of polyubiquitinated proteins to the proteasome for degradation [38], and it has also been reported to interact with mutant SOD1 in cultured cells [14]. Using two anti-p62 antibodies, staining of sections from ALS patients and controls failed to show any nuclear immunoreactivity in glial cells and motoneurons (data not shown). However, all ALS patients and 8 of the 47 controls had p62 staining in the cytoplasm, in the shape of punctuate and skein-like inclusions, as has been described before [32]. The cytoplasmic p62 aggregates showed co-localization when double-labeling was performed with ubiquitin, but no co-localization of misfolded SOD1 and p62 was seen (Online Resource, Fig. S5).
Discussion
The main new finding in this study is that misfolded SOD1 is present as small aggregates in the nuclei of glial cells of spinal cord tissue from ALS patients. These aggregates were found in ALS patients carrying SOD1 mutations as well as in sporadic and familial ALS patients lacking such mutations. Nuclear staining of misfolded SOD1 in glial cells was found in 59 of 60 ALS patients investigated. Only 4 of the 46 non-neurological and neurodegenerative controls had misSOD1 staining, but it was sparse and not seen at all levels of the spinal cord (Table 1). Since the vast majority of control cases lacked staining, the misSOD1 staining seen in ALS cases is unlikely to represent post-mortem changes, which strongly suggests that the finding was related to ALS pathology. The glial cells containing misfolded SOD1 in the nucleus were mostly astrocytes but some microglial and oligodendroglial nuclei also stained positive for misfolded SOD1 (Fig. 3). Misfolded SOD1 was occasionally found in the nuclei of motoneurons.
Many histopathological studies which included staining for SOD1, have been carried out before on ventral horns from ALS patients with and without SOD1 mutations. It could appear curious that the glial nuclear staining not has been observed before. The likely explanation is that in most cases antibodies raised against whole human SOD1 have been used. Such antibodies react avidly with both native and denatured SOD1 [12], and the minute amounts of misfolded SOD1 will thus be masked by the staining of the abundant native SOD1 in the cells. Other studies using monoclonal conformation-specific antibodies directed towards peptides in the SOD1dimer interface or in the unfolded beta-barrel have failed to show misSOD1 staining in patients with sporadic disease or non-SOD1 mediated FALS [28, 30]. A possible explanation for this could be that SOD1 in sporadic disease might have different misfolded conformation/-s compared to SOD1-mediated disease. Monoclonal peptide antibodies with only one specific antigen determinant might fail to detect other misfolded conformations, whereas polyclonal antibodies with several specific antigenic determinants could detect different misfolded confirmations. Finally, the antigen retrieval could have differed. If it is too strongly denaturing, extensive misfolding of the background native SOD1 might occur, hampering detection of the misfolded SOD1 present in the tissue.
Our finding that misfolded SOD1 is present in glial cell nuclei of FALS patients carrying SOD1 mutations as well as in ALS patients lacking such mutations support the notion of a pathological interplay between neurons and glial cells. It raises the question of how these non-motoneuron abnormalities might contribute to motoneuron degeneration and disease progression. Studies using transgenic ALS mice have shown aggregates of mutant SOD1 in astrocytes, microglia, and oligodendrocytes [7, 17, 20]. Regarding studies on human post-mortem tissue, cytosolic inclusions in spinal cord astrocytes, intensely stained with an antibody to SOD1, have been observed in two long-term surviving FALS patients carrying SOD1 mutations [27]. In the present study, all nine FALS patients carrying SOD1 mutations had staining of mutant SOD1 in glial cells, although the staining of misfolded SOD1 was primarily seen in the nuclei.
When using the mutant-specific G127X Ra 123–132 SOD1 ab, approximately 25% of the glial cell nuclei in both FALS patients with the G127X SOD1 mutant were stained, whereas using the Chi 131–153 ab, (raised to a peptide sequence that is absent in the G127X-mutated SOD1 protein) also gave staining of misfolded SOD1 in glial cell nuclei in the G127X patients. Performing double-staining with the two antibodies yielded mostly separate aggregates of misSOD1 staining and no co-localization was seen (Online Resource, Fig. S3a–c). This indicates that misfolded wild-type SOD1 protein may participate in the disease process even in patients carrying mutations.
Wild-type native SOD1 is normally located in the cytosol, the intermembrane space of mitochondria, and in the nucleus [44]. Bidirectional movement of small proteins occurs via passive diffusion through the nuclear pore complex, whereas proteins larger than about 40 kDa generally require specific transportation [41]. The size of the native SOD1 dimer is 32 kDa, and it should, therefore, be able to move freely between the nucleus and the cytosol. Accordingly, Chang et al. [8] found the native SOD1 concentration in the nucleus to be at least half that in the cytoplasm. In transgenic ALS models carrying mutant human SOD1s, misfolded SOD1 lacking the stabilizing intrasubunit C57-C146 disulfide bond is enriched in the susceptible spinal cord [48]. Such disulfide-reduced SOD1 readily forms aggregates in vitro [9, 13] and is the major component of SOD1 aggregates present in symptomatic transgenic mice [3, 26]. Misfolded disulfide-reduced SOD1 is consequently a likely source of the aggregates/inclusions of SOD1 observed in the present study. In glial cells, the aggregation mainly appears to take place in the nuclei whereas in neurons it is more prominent in the soma. Unlike dimeric and monomeric SOD1, aggregated SOD1 will be trapped and potentially accumulated in the nuclei. It is possible that both the nucleus and the cytoplasm contain targets that are vulnerable to misfolded SOD1, but that the cytoplasm of glial cells may be better equipped to neutralize or degrade the proteotoxic agent. Autophagy is the major mechanism for degradation of aggregates [18, 37] and the absence of this process in nuclei might contribute to the staining pattern of misfolded SOD1 observed. There is no evidence for extensive glial cell death in ALS. This suggests that any harmful effects of the misfolded SOD1 would not lead to glial death, but rather to activation causing damage to motoneurons. Whether the nuclear aggregates are cytotoxic, or merely terminal markers of toxic soluble misfolded disulfide-reduced SOD1 (or both species essentially innocent) can only be speculated on at present. Misfolded SOD1 may be toxic by exposing internal structures that interact with essential nuclear factors, or it may aggregate with such factors, or (as aggregates) it may physically block processes in the nuclei. Mutant SOD1 has been reported to have both RNA [15] and DNA-binding capacity [21], indicating the possibility of direct interaction with polynucleotides. In this context, it is interesting to note that several proteins found to be mutated in motor neuron diseases, such as TDP-43, FUS, angiogenin, and senataxin, can influence gene transcription and RNA turnover [40]. More details on TDP-43 and SOD1 are reported in Electronic Supplementary Material.
In neurodegenerative conditions, such as Alzheimer’s and Parkinson’s diseases, proteins found to be mutated in some familial patients (amyloid precursor protein/β-amyloid, α-synuclein) are generally assumed to be key players in the pathogenesis in sporadic patients. Immunohistochemical results constitute the strongest evidence for these suppositions. Here we have shown that inclusions of the misfolded SOD1 are regularly present in glial nuclei, both in ALS patients carrying SOD1 mutations and in sporadic and familial cases lacking such mutations. The finding expands on the previous demonstration of SOD1 inclusions in the soma of motoneurons in ALS patients lacking SOD1 mutations [5, 12]. Together, these studies suggest that misfolded SOD1 is generally involved in the pathogenesis of ALS.
As with SOD1, mutations in two other proteins, TDP-43 and FUS, have been found to cause ALS with a phenotype spectrum similar to that seen in sporadic disease. Inclusions containing TDP-43 are found in the soma of motoneurons both in carriers of TDP-43 mutations and in sporadic patients [25, 39] but not in carriers of SOD1 mutations [31] and FUS mutations [42]. Based on this discrepancy, it has been suggested that the pathogenesis of ALS caused by mutant SOD1s is different from that in sporadic ALS and ALS provoked by TDP-43 mutations. Our present findings and the findings of Bosco et al. [5] contradict this proposition even though no details on TDP-43 or FUS were provided in the latter study. Regarding involvement of the proteins found mutated in FALS pedigrees in sporadic ALS, these proteins possibly occupy different steps or occur in parallel routes in pathogenic chains of events. The fact that intranuclear glial and motoneuron inclusions were seen in the two SBMA patients as well as in the familial non-SOD1 patients suggests that SOD1 might be involved in downstream events even in motoneuron disease induced by mutations in other genes [12].
In summary, our observation that misfolded SOD1 is present in the nucleus of motoneurons and astrocytes also implicates the nucleus as a potential site of SOD1 toxicity. More studies are needed to clarify the distribution of misfolded SOD1 in non-neuronal cells and to explore in more detail the modes of SOD1 toxicity at different sites.
References
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH (2006) Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 103:16021–16026
Bergemalm D, Forsberg K, Srivastava V, Graffmo KS, Andersen PM, Brannstrom T, Wingsle G, Marklund SL (2010) Superoxide dismutase-1 and other proteins in inclusions from transgenic amyotrophic lateral sclerosis model mice. J Neurochem 114:408–418
Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312:1389–1392
Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien JP, Brady ST, Brown RH Jr (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 13:1396–1403
Brooks BR, Miller RG, Swash M, Munsat TL (2000) El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 1:293–299
Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18:327–338
Chang LY, Slot JW, Geuze HJ, Crapo JD (1988) Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J Cell Biol 107:2169–2179
Chattopadhyay M, Durazo A, Sohn SH, Strong CD, Gralla EB, Whitelegge JP, Valentine JS (2008) Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc Natl Acad Sci USA 105:18663–18668
Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH Jr, Julien JP, Goldstein LS, Cleveland DW (2003) Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302:113–117
Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, Lukas T, Dal Canto MC, O’Halloran TV, Siddique T (2006) Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci USA 103:7142–7147
Forsberg K, Jonsson PA, Andersen PM, Bergemalm D, Graffmo KS, Hultdin M, Jacobsson J, Rosquist R, Marklund SL, Brannstrom T (2010) Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE 5:e11552
Furukawa Y, Kaneko K, Yamanaka K, O’Halloran TV, Nukina N (2008) Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J Biol Chem 283:24167–24176
Gal J, Strom AL, Kwinter DM, Kilty R, Zhang J, Shi P, Fu W, Wooten MW, Zhu H (2009) Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem 111:1062–1073
Ge WW, Wen W, Strong W, Leystra-Lantz C, Strong MJ (2005) Mutant copper-zinc superoxide dismutase binds to and destabilizes human low molecular weight neurofilament mRNA. J Biol Chem 280:118–124
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX et al (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264:1772–1775
Gurney ME (1997) Transgenic animal models of familial amyotrophic lateral sclerosis. J Neurol 244(Suppl 2):S15–S20
Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, Kopito RR (2005) Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci USA 102:13135–13140
Jaarsma D, Haasdijk ED, Grashorn JAC, Hawkins R, Van Duijn W, Verspaget HW, London J, Holstege JC (2000) Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motorneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiology of Disease 7:623–643
Jaarsma D, Teuling E, Haasdijk ED, De Zeeuw CI, Hoogenraad CC (2008) Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J Neurosci 28:2075–2088
Jiang W, Han Y, Zhou R, Zhang L, Liu C (2007) DNA is a template for accelerating the aggregation of copper, zinc superoxide dismutase. Biochemistry 46:5911–5923
Jonsson PA, Ernhill K, Andersen PM, Bergemalm D, Brannstrom T, Gredal O, Nilsson P, Marklund SL (2004) Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain 127:73–88
Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Lindberg M, Oliveberg M, Marklund SL (2006) Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain 129:451–464
Jonsson PA, Graffmo KS, Brannstrom T, Nilsson P, Andersen PM, Marklund SL (2006) Motor neuron disease in mice expressing the wild type-like D90A mutant superoxide dismutase-1. J Neuropathol Exp Neurol 65:1126–1136
Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574
Karch CM, Prudencio M, Winkler DD, Hart PJ, Borchelt DR (2009) Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc Natl Acad Sci USA 106:7774–7779
Kato S, Hayashi H, Nakashima K, Nanba E, Kato M, Hirano A, Nakano I, Asayama K, Ohama E (1997) Pathological characterization of astrocytic hyaline inclusions in familial amyotrophic lateral sclerosis. Am J Pathol 151:611–620
Kerman A, Liu HN, Croul S, Bilbao J, Rogaeva E, Zinman L, Robertson J, Chakrabartty A (2010) Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol 119:335–344
Kinoshita Y, Ito H, Hirano A, Fujita K, Wate R, Nakamura M, Kaneko S, Nakano S, Kusaka H (2009) Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 68:1184–1192
Liu HN, Sanelli T, Horne P, Pioro EP, Strong MJ, Rogaeva E, Bilbao J, Zinman L, Robertson J (2009) Lack of evidence of monomer/misfolded superoxide dismutase-1 in sporadic amyotrophic lateral sclerosis. Ann Neurol 66:75–80
Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VM, Trojanowski JQ (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434
Mizuno Y, Amari M, Takatama M, Aizawa H, Mihara B, Okamoto K (2006) Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J Neurol Sci 249:13–18
Pardo CA, Xu Z, Borchelt DR, Price DL, Sisodia SS, Cleveland DW (1995) Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc Natl Acad Sci USA 92:954–958
Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi XF, Crow JP, Cashman NR, Kondejewski LH, Chakrabartty A (2002) Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem 277:47551–47556
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Rouleau GA, Clark AW, Rooke K, Pramatarova A, Krizus A, Suchowersky O, Julien JP, Figlewicz D (1996) SOD1 mutation is associated with accumulation of neurofilaments in amyotrophic lateral sclerosis. Ann Neurol 39:128–131
Rubinsztein DC (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443:780–786
Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW (2004) Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 24:8055–8068
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672
Strong MJ (2010) The evidence for altered RNA metabolism in amyotrophic lateral sclerosis (ALS). J Neurol Sci 288:1–12
Terry LJ, Shows EB, Wente SR (2007) Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science 318:1412–1416
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Wang L, Sharma K, Deng HX, Siddique T, Grisotti G, Liu E, Roos RP (2008) Restricted expression of mutant SOD1 in spinal motor neurons and interneurons induces motor neuron pathology. Neurobiol Dis 29:400–408
Weisiger RA, Fridovich I (1973) Superoxide dismutase. Organelle specificity. J Biol Chem 248:3582–3592
Wroe R, Wai-Ling Butler A, Andersen PM, Powell JF, Al-Chalabi A (2008) ALSOD: the amyotrophic lateral sclerosis online database. Amyotroph Lateral Scler 9:249–250
Yamanaka K, Boillee S, Roberts EA, Garcia ML, McAlonis-Downes M, Mikse OR, Cleveland DW, Goldstein LS (2008) Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc Natl Acad Sci USA 105:7594–7599
Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253
Zetterstrom P, Stewart HG, Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Oliveberg M, Marklund SL (2007) Soluble misfolded subfractions of mutant superoxide dismutase-1 s are enriched in spinal cords throughout life in murine ALS models. Proc Natl Acad Sci USA 104:14157–14162
Acknowledgments
We thank Eva Bern, Ingalis Fransson, Karin Hjertkvist, Dan Nylund, Ulla-Stina Spetz, Frode Sörli and Agneta Öberg for expert technical assistance and dr Ole Gredal and the ethic committee at the University of Copenhagen. This work was supported by the Swedish Science Council (Grants nr: 2008:2786; 2009:3548; 2009:4942), the Swedish Brain Fund/Hållsten Fund, The Swedish Medical Society/Björklund Fund for ALS Research, King Gustaf and Queen Victoria Fund, The Swedish Association for individuals with Neurological Disabilities, the Torsten and Ragnar Söderbergs Foundations, the Västerbotten County Council, and the Research Fund of the Medical Faculty, Umeå University.
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Forsberg, K., Andersen, P.M., Marklund, S.L. et al. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol 121, 623–634 (2011). https://doi.org/10.1007/s00401-011-0805-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-011-0805-3