
Abstract
The synthesis, characterization, self-assembly, and gel formation of poly(γ-benzyl-l-glutamate) (PBLG) in a molecular weight range from ca. 7,000–100,000 g/mol and with narrow molecular weight distribution are described. The PBLG is synthesized by the nickel-mediated ring-opening polymerization and is characterized by size-exclusion chromatography coupled with multiple-angle laser light scattering, NMR, and Fourier transform infrared spectroscopy. The self-assembly and thermoreversible gel formation in the helicogenic solvent toluene is investigated by transmission electron microscopy, atomic force microscopy, small-angle X-ray scattering, and synchrotron powder X-ray diffraction. At concentrations significantly below the minimum gelation concentration, spherical aggregates are observed. At higher concentrations, gels are formed, which show a 3D network structure composed of nanofibers. The proposed self-assembly mechanism is based on a distorted hexagonal packing of PBLG helices parallel to the axis of the nanofiber. The gel network forms due to branching and rejoining of bundles of PBLG nanofibers. The network exhibits uniform domains with a length of 200 ± 42 nm composed of densely packed PBLG helices.

Similar content being viewed by others

Introduction
The elucidation of thermoreversible gelation of rod-like polymers has been a challenging field of research for decades [1–10]. There are several different approaches for the explanation of thermoreversible gelation of rod-like polymers. Nandi and coworkers [11] summarize the most commonly discussed mechanisms as (1) microphase separation [1], (2) entanglement formation [12], (3) rod jamming [13], and (4) glass transition [14]. Flexible polymers can easily form domains with crystalline or semicrystalline structures. These domains act as cross-links to stabilize a gel network. It is less clear how rod-like polymers can participate in intermolecular physical cross-links. These cross-links are the prerequisite for the formation of a thermoreversible gel network. However, rod-like polymers often form thermoreversible gels [1, 15, 16]. Many groups have shown that rod-like polymers have the ability to aggregate into microfibrils or nanofibers under specific solvent and temperature conditions [7, 8, 10, 17]. These nanofibers can be considered as supramolecular, flexible aggregates, whose formation is reversible. It is assumed that the mechanism of gelation by the formation of intermolecular cross-links is also applicable to rod-like polymers. First, they self-assemble into nanofibers, and then, these nanofibers, which are comparable to coil polymers due to their flexibility, form crystalline domains acting as cross-links.
The investigation of the gel formation of rod-like polymers is not only of academic interest but also of great importance for applications. For example, high-performance fibers, which are composed of a microfibrillar network, are spun from solutions of rigid polymers [10]. The manipulation of the microfibrillar structure by controlling its formation kinetics is a current challenge [18]. Furthermore, rigid polymer gels can be used to fabricate new membranes or lightweight foam [4], where structural control is important [10].
Poly(γ-benzyl-l-glutamate) (PBLG) is a synthetic polypeptide that can adopt a rigid α-helical conformation depending on its molecular weight both in solution and in the solid state [19, 20]. The intrinsic rigidity of PBLG in helicogenic solvents is due to stable intramolecular hydrogen bonding [21]. PBLG forms aggregates in dilute solutions of some helicogenic solvents such as toluene and exhibits thermoreversible gelation above critical concentrations [1, 2, 4, 6, 8, 22, 23]. For the mechanism of the gel formation of PBLG in helicogenic solvents like toluene, a variety of models exist. However, a consistent mechanism, which includes all observations, is still not established. Cohen and coworkers [10] provided an overview of the mechanisms discussed in literature. Early models associate the gelation process, which occurs when isotropic solutions of PBLG are cooled, with liquid–liquid phase separation by “spinodal decomposition” [1, 24, 25]. This model is supported for hydroxypropyl cellulose in water [26], PBLG in DMF/H2O [27], and PBLG–toluene gels under some circumstances [28]. The influence of the parameters like temperature, concentration, solvent, etc., on the gelation can be understood in terms of a competition between aggregation phenomena and phase separation [8, 24]. An alternative gelation mechanism of PBLG solutions is based on “nucleation and growth” of a crystalline phase [4, 15, 29]. Measurements of enthalpic endotherms during gel melting support the existence of crystals in the gel structure [4, 30]. Cohen and coworkers [31] propose the basis for the formation of gel microfibrils to be the formation of crystal solvate phases. The PBLG cocrystallizes with solvent molecules. Jackson et al. [4] and Horton et al. [5] suggest a mixed mechanism for the gelation of PBLG solutions. A liquid–liquid phase separation, in which separation occurs either by “spinodal decomposition” or “nucleation and growth,” is followed by a crystallization in the concentrated phase.
Shukla [6] gives a descriptive explanation for the formation of gels starting from dilute solutions of PBLG in benzyl alcohol: At elevated temperatures, PBLG chains are randomly dispersed and do not interact with each other. Shukla does not take into account the end-to-end aggregation of PBLG chains observed by Chakrabarti et al. [32] even in hot toluene solutions. This kind of aggregation phenomenon may occur in benzyl alcohol as well. When the temperature is decreased, Shukla suggests that bundles of three or four PBLG chains are formed due to stacking of the side-chain benzene rings (π–π interactions). At reduced temperature, the intermolecular interactions between PBLG chains are strong enough to maintain the bundle structure. The bundles are assumed to be 2.0–2.5 nm in diameter. As the temperature is further decreased, these bundles aggregate and form fibers, which may contain five to ten bundles and are about 10 nm in diameter. Shukla comments that the exact nature of the intermolecular forces causing the aggregation is not known. However, for an infinite network to be formed, these fibers need to be cross-linked. The formation of cross-links takes places by the branching and rejoining of different sheaf-like aggregates throughout the solution, resulting in an interconnected fibrillar morphology [32]. The same mechanism of branching and rejoining has been proposed by Tadmor et al. from investigation of the gelation kinetics by small-angle neutron scattering [10].
In this article, the synthesis of a series of PBLG homopolymers is presented along with the investigation of their thermoreversible gel formation in the helicogenic solvent toluene. PBLG nanofibers form at concentrations well below the minimum gelation concentration. In this state, individual nanofibers can be visualized and analyzed to determine their lateral dimensions by transmission electron microscopy (TEM) and atomic force microscopy (AFM). Small-angle x-ray scattering and synchrotron powder X-ray diffraction (SAXS and SyPXRD) are applied to elucidate the long-range order of the nanofibers and the gel network structure.
Experimental section
Materials
All solvents were dried and distilled using standard procedures [33] and, if necessary, degassed by a “freeze–pump–thaw” procedure. γ-Benzyl-l-glutamic acid (BLG, >99 %, Fluka) and phosgene solution (∼20 % in toluene, purum, Fluka) were used as received. γ-Benzyl-l-glutamate-N-carboxyanhydride (BLG–NCA) was synthesized as previously described [34]. The preparation of the precursor for the bifunctional initiator, l-leucine bromo isobutyric acid amino ethylester, was also performed as previously described [35].
Methods
SEC–MALLS
Molecular weights and molecular weight distributions were determined by size-exclusion chromatography coupled with multiple-angle laser light scattering (SEC–MALLS) in DMF (membrane filtered and degassed) containing LiBr (0.1 mol/L) on two PSS GRAM columns (1,000 Å, 10 μm, 8.0 × 300 mm, PSS—Polymer Standards Service, Mainz) at 60 °C and a flow rate of 0.5 mL/min. Detection was performed with a Shodex RI-101 differential refractive index detector set at 45 °C and a TriStar MiniDawn light scattering detector from Wyatt Technology (angles at 30°, 90°, and 120°; laser wavelength, 690 nm). The refractive index increment dn/dc was measured on a BTC Brookhaven instrument using a wavelength of 620 nm. At least five samples with different concentrations ranging from 1-10 mg/mL in the same solvent as for the SEC measurements were investigated for the determination of the dn/dc value via linear regression.
TEM
TEM images were obtained on a Hitachi H-7000 microscope operated at an acceleration voltage of 100 kV. For samples below the minimum gelation concentration (C gel), the specimens were prepared by placing one drop of the clear solution onto the carbon-coated copper grid, removing the solvent with a tissue and air-drying the sample for 1 min with an airflow. For samples at or above C gel, the TEM specimens were prepared by gently placing the TEM grid on the fraction of organogel. The TEM grid was removed after a few seconds and air-dried for 1 min. The specimens were measured 30 min after their preparation. For the images, the gray scale was inverted by software to enhance visibility of nanofibers and facilitate the comparison with AFM images. For the analysis of the width of nanofibers, the software ImageJ 1.42q (copyright by Wayne Rasband, National Institutes of Health, USA; http://rsb.info.nih.gov/ij) was used.
AFM
AFM was performed on an extended multimode Nanoscope IIIa (Digital Instruments, now Veeco, Santa Barbara, CA) in the Tapping Mode™. The sample preparation for AFM measurement was identical to the preparation of the TEM specimens. Freshly cleaved mica was used as a substrate. For the data analysis, the software Gwyddion 2.19 (http://gwyddion.net/) was used. All images were leveled by mean plane subtraction, and the minimum value was set to zero.
FT-IR spectroscopy
Fourier transform infrared spectroscopy (FT-IR) spectra were measured on the instrument FT-IR-Equinox IFS 55 from Bruker. Polymers were dissolved in chloroform and measured as a film on a KBr pellet relative to a pure KBr pellet (KBr for IR spectroscopy, Aldrich, ≥99 %). Analysis of IR spectra was performed with OPUS 4.0 from Bruker.
NMR
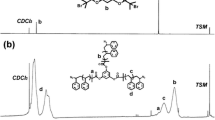
1H- and 13C-NMR spectra were recorded on the instrument DPX 400 from Bruker. Calibration of chemical shifts was performed relative to TMS. For monomers, the concentration was ca. 10 mg/0.7 mL in CDCl3 and for polymers ca. 30 mg/0.7 mL in CDCl3. Abbreviations for multiplicity are as follows: s (single), d (double), t (triple), and m (multiple). Multiplicity of 13C-NMR signals was determined with DEPT technique at a spin angle of 135°.
SAXS
SAXS measurements were performed with a SAXSess camera (Anton Paar, Austria) attached to a laboratory X-ray generator (PW3830, PANanalytical) operated with a fine-focus glass X-ray tube at 40 kV and 50 mA (CuKα, λ = 0.1542 nm). Samples were measured in the dry state between Mylar films. The scattering vector, which is defined in terms of the scattering angle θ and the wavelength of the radiation λ, is q = 4π/λ sin(θ/2). SAXS data were recorded in a q range of 0.04 to 5.0 nm−1 with a CCD detection system (Anton Paar, Austria). The 2D intensity data were converted to 1D data and deconvoluted using the software SAXSQuant (Anton Paar, Austria). Data were fitted using Igor Pro 6.0.4 (Wavemetrics) and a Teubner-Strey [36, 37] model as implemented in the Irena Package [38] version 2.36.
SyPXRD
Samples were measured as a sandwich between adhesive tape at the μSpot beamline [39] at BESSY-II, using a wavelength of 1.5406 Å. Silicon was used as external standard, and the 2D data were converted using Fit2D [40].
Synthesis
Synthesis of nickel (amido amidate l-leucine bromo isobutyric acid amino ethylester-phenanthroline) [Ni(phen)]
Under dry and oxygen-free conditions, 496 mg (1.8 mmol, 1 eq.) Ni(COD)2 was suspended in 35 mL of dried DMF, and 329 mg (1.8 mmol, 1 eq.) 1,10-phenanthroline was dissolved in 10 mL of dried DMF. The dissolved phenanthroline was transferred with a N2-purged syringe to the Ni(COD)2 suspension, and an immediate change to a dark green color was observed. After stirring for 2 h at room temperature, 737 mg (1.8 mmol, 1 eq.) l-leucine bromo isobutyric acid amino ethylester (synthesis of this precursor is described by Steig et al. [35]), dissolved in 10 mL dried DMF, was added via a N2-purged syringe. The solution was first stirred for 24 h at room temperature until it turned green and subsequently for another 24 h at 50 °C. After allowing the reaction mixture to cool down to room temperature, 190 mL of diethyl ether was added, and a finely dispersed powder precipitated. The product was isolated by pouring the reaction mixture over a reverse frit and washing the fine powder two times with 10 mL of diethyl ether and four times with 10 mL of THF. The green powder was dried in vacuum for at least 5 h and was stored under nitrogen. Yield: 537 mg (0.965 mmol, 54 %), EA: N: 8.78 (9.97); C: 43.93 (51.28); H: 4.30 (5.56); Br: 15.45 (14.21). FT-IR (KBr pellet, cm−1): 3,399 (NH valence), 3,056 (CH valence, aromatic), 2,957 (CH valence, aliphatic), 1,723 (ester carbonyl vibration), 1,661 (C=O valence, amide), 1,585 (C=O, amidate), 854/732 (out of plane, substituted aromatic). Because of the paramagnetism of nickel, no NMR spectra could be recorded.
Synthesis of nickel (amido amidate l-leucine bromo isobutyric acid amino ethylester-bis-(diethylphosphinoethane)) [Ni(depe)]
Under dry and oxygen-free conditions 900 mg (3.3 mmol, 1 eq.) Ni(COD)2 was suspended in 60 mL of dried THF. and 760 μL (3.3 mmol, 1 eq.) bis-(diethylphosphinoethane) [depe] was dissolved in 15 mL of dried THF. The dissolved depe was transferred with a N2-purged syringe to the Ni(COD)2 suspension, and an immediate change to a dark-orange color was observed. After stirring for 2 h at room temperature, 1.31 g (3.3 mmol, 1 eq.) l-leucine bromo isobutyric acid amino ethylester (synthesis of this precursor is described by Steig et al. [35]), dissolved in 15 mL of dried THF, was added via a N2-purged syringe. The solution was stirred for 24 h at 80 °C. After allowing the reaction mixture to cool down to room temperature, the reaction mixture was poured into 200 mL of petroleum ether (PE), freshly dried over sodium, and a finely dispersed brown–orange powder precipitated. The product was isolated by applying a filtration with a reverse frit and washing the fine powder four times with 10 mL of freshly dried PE each. The brown–orange powder was dried in vacuum for 1 day and was stored under nitrogen. Yield: 987 mg (1.7 mmol, 51 %). EA: N: 4.79 (4.76); C: 41.93 (44.92); H: 7.19 (8.05); Br: 12.38 (13.58). FT-IR [KBr pellet, cm−1]: 3,293 (NH valence), 2,960 (CH valence, aliphatic), 1,722 (ester carbonyl vibration), 1,625 (C=O valence, amide), 1,583 (C=O, amidate). Because of the paramagnetism of nickel, no NMR spectra could be recorded.
Synthesis of PBLG homopolymer
BLG–NCA, 1.5 g (5.7 mmol, 1 eq.) (see Supporting info), was dissolved in a dry Schlenk flask in 11 mL of dried DMF, and 8.5 mg (0.015 mmol, 0.003 eq., [M]/[I] = 380) Ni(phen) or 8.8 mg (0.015 mmol, 0.003 eq., [M]/[I] = 380) Ni(depe) was added. The solution was then stirred under a nitrogen atmosphere for 24 h. The polymerization was stopped by opening the reaction flask to air under ice-cooling. Subsequently, the polymer was isolated by precipitation. For this purpose, the reaction mixture was diluted with THF in case the viscosity was too high. The polymer was precipitated into ice-cooled methanol with HCl (4 mmol/L) added to hydrolyze the reactive nickel end group. The resulting powder was isolated by filtration and dried under vacuum at room temperature overnight. The polymer was purified by two reprecipitation steps from THF. Yields and data for the characterization via SEC–MALLS are given in Table 1. Additional analytical data are presented in the Supporting information.
Preparation of polymer sols and gels
After dissolving the polymer under vigorous stirring in toluene at 70 °C for 30–60 min, the solution was allowed to cool to room temperature. The gel formed 5–10 min after removing the sample from heating. If the polymer concentration was too low, gel particles which floated in the toluene formed after cooling the solution to room temperature. The viscosity of the solution did not increase significantly compared to pure toluene. In some cases, a gel was formed, but the vial inversion test was negative, indicating that the gel was not self-supportive. The nonstable gel collapsed into a liquid and gel pieces. When the polymer concentration was at C gel, the vial inversion test was positive indicating that a self-supportive gel was formed.
Results and discussion
Synthesis and characterization of PBLG homopolymers
The nickel-mediated NCA polymerization, introduced by Deming and coworkers [41–46], was applied to synthesize the PBLG homopolymers. The polymerization was initiated by a nickel-amido-amidate complex, either Ni(phen) or Ni(depe), in DMF and conducted for 24 h at room temperature (Scheme 1). We used bifunctional initiators to prepare PBLG having a well-defined end group, as has been previously shown by MALDI-ToF [35]. Further studies would be possible using the endgroup for initiating the polymerization of, e.g., a methacrylate polymer, yielding rod–coil block copolymers [35], which should show a higher degree of complexity in their aggregation behavior. However, here, we focus on the properties of PBLG homopolymers.

Reaction scheme for the synthesis of PBLG via the bifunctional initiator Ni(phen) or Ni(depe)
The PBLGs were characterized by means of SEC-MALLS, 1H- and 13C-NMR spectroscopy and FT-IR (see the Supporting information for more details). Number average molecular weight M n,SEC is determined by SEC–MALLS with dn/dc (PBLG) = 0.122 mL/g (45 °C). Table 1 shows the conversion, molecular weight, and polydispersity index (PDI) for selected PBLGs.
The conversions were in the range of 39–70 %, molecular weights in the range of 6,500 to 97,000 g/mol, and PDIs in the range of 1.13 to 1.55. The Ni(depe) initiator has the advantage that PBLG homopolymers with a narrower molecular weight distribution (PDI ca. 1.1) compared to PBLG homopolymers synthesized by Ni(phen) (PDI ca. 1.4) were accessible. However, for preparing PBLG with higher molecular weight, Ni(phen) is suited better. Utilizing both nickel-based initiators, molecular weights from ca. 7,000 to ca. 100,000 g/mol can be prepared. High molecular weight polypeptides are available using amine or methoxide initiators [21, 47]; however, in this case, the polymerization proceeds via the so-called “activated monomer” mechanism, and broader molecular weight distributions result [48]. Employing primary amines as initiators, the polymerization proceeds via the “amine mechanism” [48], and narrow molecular weight distributions can be obtained, but typically, the molecular weight is rather limited [49]. Therefore, a broad molecular weight range as prepared here by transition metal initiators is hardly accessible by the amine-initiated NCA polymerization. For example, Kuo et al. [50] synthesized PBLG blocks by utilizing the amine-initiated NCA polymerization in the range of only 6,000–15,000 g/mol (M n determined by 1H-NMR) requiring a reaction time of 5 days at room temperature.
The α-helical character of selected PBLG samples was investigated by FT-IR (see Supporting information, SI Fig. 2). The two PBLG samples PBLG30 and PBLG338 show the same bands at 1,653 and 1,548 cm−1. According to Block [19], polypeptides in the α-helical conformation show the amide I band at 1,650 cm−1 and the amide II band at 1,546 cm−1, whereas polypeptides adapting a β-sheet conformation show the amide bands at 1,630 and 1,530 cm−1. Thus, it can be concluded that the PBLG samples exclusively show the α-helical conformation.
Gel formation of PBLG in toluene
The ability of different PBLG homopolymers to form thermoreversible gels in the helicogenic solvent toluene was investigated. PBLG becomes soluble in toluene when heated to ca. 70 °C. When the clear solution is cooled, a gel is formed. As a characteristic and easily accessible parameter for this type of gel, the minimum gelation concentration (C gel) is determined by varying the concentration and performing the vial inversion test (Fig. 1).

Vial inversion test for three different polymer concentrations in toluene. For the first concentration, the gel is not stable and collapses into gel pieces and toluene. For the other two concentrations, stable gels are formed
Table 2 shows the molecular weight, PDI, helix length, and C gel value for selected PBLGs. With increasing molecular weight or PBLG α-helix length, the C gel of PBLG was significantly reduced. The gel formation of PBLG is thermoreversible, with a gel-sol transition temperature (T gel) in the range of 48–53 °C. The higher the molecular weight of the PBLG and the higher the concentration of the polymer, the higher T gel.
Investigation of the morphology for solutions of PBLG at C << C gel in toluene
The self-assembled structures formed in PBLG sols well below C gel were investigated using TEM and AFM. After cooling a highly diluted solution of PBLG in toluene from ca. 70 °C to RT, it was observed by the naked eye that small aggregates had formed. The viscosity of the toluene did not visibly increase, and the solution remained transparent. The TEM image of a dried sample taken from a 0.05 wt% solution of PBLG338 in toluene (Fig. 2a) reveals almost spherical aggregates, which stick together.

a TEM image of dried aggregates from a 0.05-wt% solution of PBLG338 in toluene. b TEM image of nanofibers from a 0.05-wt% solution of PBLG338 in toluene. Arrows indicate the diminution of a thick bundle of nanofibers. c AFM height image of nanofibers from a 0.05-wt% solution of PBLG338 in toluene. The white bar represents a scale of 1 μm. d AFM height profile from the cross section in Fig. 2c
Thin nanofibers radiate from these aggregates; thus, they can be described as spheres with tentacles. The structures formed between adjacent spheres are most likely dense aggregates of entangled nanofibers. Figure 2b shows a higher magnification TEM image from the periphery of a sphere, where nanofibers of various widths can be seen. The arrows indicate a randomly chosen nanofiber, whose width decreases along the axis. This observation indicates that a thicker nanofiber is composed of a bundle of thinner nanofibers, which end at some point or branch. The thinnest nanofibers present in the TEM image have an average width of ca. 24.5 ± 3.4 nm. The histogram is shown in Fig. 3.

Histogram of 50 width measurements of the thinnest nanofibers of a 0.05-wt% solution of PBLG338 in toluene from TEM image (Fig. 4b). The average width of the thinnest nanofibers is 24.5 ± 3.4 nm
Figure 2c shows the AFM height image of nanofibers from a 0.05-wt% solution of PBLG338. The white dashed cross section covers six prominent nanofibers. Figure 2d presents the height profile of these nanofibers, which exhibit a height in the range of 4–9 nm. The determination of the height of the thinnest nanofibers in the background in Fig. 2c is not reliable due to limitations of AFM, which cannot resolve the dimensions of “buried” structures accurately. However, the nanofibers might consist of a uniformly sized basic fiber, which is capable of forming fiber bundles. For example, in Fig. 2c, it can be seen that for nanofiber 3 two strands merge, and then, the nanofiber branches into thinner nanofibers. According to the widths in the range of ca. 21–28 nm and heights in the range of 4–9 nm, it can be speculated that the nanofibers consist of 14–18 PBLG helices densely packed side by side and two to six PBLG helices stacked assuming a PBLG helix diameter of 1.5 nm [19, 51].
Figure 4 shows detailed AFM images of spherical aggregates and bundles of nanofibers from a dried sample of a 0.05-wt% solution of PBLG338 in toluene on a freshly cleaved mica surface. The question arises why these nanofibers are formed when a hot solution of PBLG in toluene is allowed to cool down. Miller and coworkers [32] discovered that PBLG aggregates end to end even in hot toluene. Figure 5 shows a schematic illustration of the proposed transition of dissolved PBLG in hot toluene to PBLG spheres with tentacles at PBLG concentrations below C gel and self-assembled PBLG networks at PBLG concentrations at or above C gel.

AFM images of dried aggregates and bundles of nanofibers from a 0.05-wt% solution of PBLG338 in toluene. a Spherical aggregates fused together. b Spheres sticking together via entanglements of their “nanofiber corona.” c Strands of nanofibers surrounded by very thin nanofibers. d Detailed image of strands of nanofibers

a Schematic illustration of proposed structures for supramolecular chains of end-to-end aggregated PBLG helices in hot toluene; b formation of spheres upon cooling when the PBLG concentration is below C gel; and c formation of a self-supportive gel when the PBLG concentration is at or above C gel
Figure 5 (A) depicts end-to-end aggregated PBLG helices in hot toluene. These supramolecular chains of end-to-end assembled PBLG helices can be considered as precursors for the formation of nanofibers. A single nanofiber precursor would have a diameter of a PBLG α-helix (i.e., 1.5 nm) and a length of a multiple of the length of one helix. They start to aggregate side by side forming flexible nanofibers upon cooling. This side-by-side aggregation is caused by the π–π interaction of the aromatic side chains. As pointed out by Shukla et al. [6], bundles of four helices with approximately 2–2.5-nm thickness are formed as primary aggregates, while Korenaga et al. [28] suggested three helices in an aggregate. These bundles then may undergo a further level of aggregation forming nanofibers as already proposed by Izumi et al. [30], resulting in relatively uniform thickness of the nanofibers. In the case the concentration is low, the nanofibers form spherical particles with tentacles (Fig. 5, B). The formation of the spheres is most likely to reduce the surface area and thus the unfavorable interaction with the solvent. When the PBLG concentration is sufficient to form nanofibers, which can fill the whole vial, a self-supportive gel is obtained (Fig. 5, C). The concentration-dependent gel formation can be explained with the model of supramolecular PBLG chains. When the polymer concentration is at C gel, PBLG nanofibers are formed, which are long enough and the whole vial can be covered. Cross-links are due to branching points of bundles of nanofibers. To form such long nanofibers, a higher concentration is required for short PBLGs than for long PBLGs.
Investigation of PBLG gels in toluene at C ≥ C gel by TEM, SAXS, and SyPXRD
The gel structure was investigated by TEM. The TEM picture (Fig. 6a) reveals a network structure, which is composed of nanofibers that branch and rejoin. It can be seen that the network is disrupted at some points, which may be caused by electron beam damage.

a TEM image of a dried sample from a 0.3-wt% PBLG338 gel in toluene. b SAXS pattern of a dried sample from a 2-wt% PBLG338 gel in toluene. Diffraction peak at d = 1.47 nm can be attributed to the distance between the centers of adjacent PBLG helices. c SyPXRD pattern of a dried sample from a 2-wt% PBLG338 gel in toluene. The diffraction peak at 0.5 nm indicates the pitch distance in the PBLG α-helix
SAXS and SyPXRD were performed to elucidate the PBLG gel long-range order and the possible structure of PBLG bundles. SAXS data (Fig. 6b) show a clear q −4 trend and thus demonstrate a typical Porod-type behavior. This is indicative of a well-defined internal interface [36]. This interface can be visualized as a sharp interface between aligned PBLG helices and open space in between the fibers. These results are in line with the interpretation of SAXS measurements on nascent gels by Izumi et al. [30] who have suggested that the aggregates can be approximated as isolated assemblies of parallel rods. Quantitative information about this system was gained using a Teubner-Strey fitting [36, 37]. In principal, this model allows the interpretation of lamellar-like structures. In our case, the rigid PBLG rods act as structural equivalents of self-organized surfactants because of their shape and rigidity. Data fitting leads to a domain or coherence length d of ca. 200 nm and a correlation length ξ of ca. 42 nm, the latter representing some sort of standard deviation or a measure for the polydispersity of the domain length. The SAXS curve also exhibits a peak at d = 1.47 nm, which corresponds to the distance between densely packed PBLG helices [19]. In summary, SAXS data show that the gel fibers are composed of independent domains with a length of 200 ± 42 nm parallel to the fiber long axis, and the individual PBLG helices are densely packed in a hexagonal fashion (vide infra).
In order to confirm this, SyPXRD was performed on a dried sample. The image recorded on a 2D detector (Fig. 6c, inset) of a dried film (not prepared to be oriented) qualitatively shows that there is some preferential orientation, even if the effects are limited. In order to obtain more quantitative data, the image plate was integrated and the corresponding diffractogram is shown in Fig. 6c. There is a diffraction peak at d = 0.5 nm, which can be attributed to the helical pitch of an α-helical conformation [52] This confirms that PBLG338, which was found to be α-helical by FT-IR, keeps its α-helical conformation in the dried gel state. Moreover, the diffractogram shows four peaks at 2θ = 6.13°, 12.38°, 20.59°, and 25.24° (respectively 1.44, 0.72, 0.43, and 0.35 nm), which can be ordered as (100), (200), (−130/−230), and (400) reflections of PBLG in form B [53]. This PBLG form, noted as form B, was first discovered and analyzed by Tobolsky and coworkers [53]. It is a well-ordered modification consisting of PBLG helices packed on a regular 2D oblique net. This oblique net can be denoted as a distorted hexagonal lattice. SyPXRD also provides information about inter-PBLG orientation. In the PBLG form B, all senses of rotation of α-helices are randomly distributed. Figure 7 shows a model for the PBLG network based on the SAXS and SyPXRD results. The domains within the network have a length of ca. 200 nm. Within these domains, the PBLG helices are packed in a distorted hexagonal lattice. The lattice contains a mixture of antiparellel PBLG helices, which have different senses of rotation when observed from the same end group. The antiparallel arrangement of PBLG helices is also found in the self-assembly of PBLG-based rod-coil block copolymers [54].

Model for the network formation of bundles of PBLG helices
The results show that a limited crystallization is involved in the gel formation of PBLG in toluene. Uniformly sized PBLG nanofibers with a diameter of one α-helix are assumed to aggregate side by side during gel formation. By this aggregation process, crystalline domains with an average length of ca. 200 nm and a width of several PBLG helix diameters are formed. These domains act as cross-links to stabilize a 3D-PBLG network structure. Thus, the gel formation mechanism including crystallization proposed by Horton et al. [5] could be supported.
Summary
The synthesis of PBLGs in a molecular weight range from ca. 7,000–100,000 g/mol with monomodal molecular weight distributions was achieved by using a nickel-based initiator. PBLG forms thermoreversible gels in toluene in a concentration-dependent manner. The lower the molecular weight of the PBLG, the higher the minimum gelation concentration. The self-assembled structures formed at very low concentrations of PBLG in toluene were studied by means of TEM and AFM. These techniques revealed a morphology which can be described as “spheres with tentacles.” The spheres are comprised of densely aggregated nanofibers and stick together by entanglements of their “nanofiber corona.” The nanofibers are most likely a result of side-by-side merging of end-to-end aggregated PBLG helices. It is assumed that the gel is formed when the polymer concentration is high enough so that long enough nanofibers are formed, which can fill the whole vial. With the support of X-ray scattering data (SAXS and SyPXRD) of dried gels, a model for the PBLG nanofibers and network could be derived. The nanofibers consist of densely packed PBLG helices in a distorted hexagonal array aligned parallel to the axis of the fiber. Additionally, it was found that crystalline domains with a length of 200 nm act as cross-links to support the 3D-PBLG network structure.
References
Tohyama K, Miller WG (1981) Nature 289:813–814
Uematsu I, Uematsu Y (1984) In: Liquid Crystal Polymers I, p 37–73
Hill A, Donald AM (1988) Polymer 29:1426–1432
Jackson CL, Shaw MT (1990) Polymer 31:1070–1084
Horton JC, Donald AM (1991) Polymer 32:2418–2427
Shukla P (1992) Polymer 33:365–372
Cohen Y (1996) J Polym Sci B Polym Phys 34:57–64
Tipton DL, Russo PS (1996) Macromolecules 29:7402–7411
Cohen Y, Dagan A (2002) Macromolecules 28:7638–7644
Tadmor R, Khalfin RL, Cohen Y (2002) Langmuir 18:7146–7150
Malik S, Jana T, Nandi AK (2000) Macromolecules 34:275–282
Russo PS (1987) Reversible polymeric gels and related systems. American Chemical Society, Washington, DC
Edwards SF, Evans KE (1982) J Chem Soc Faraday Trans2: Mol Chem Phys 78:113–121
Ren SZ, Sorensen CM (1993) Phys Rev Lett 70:1727
Sasaki S, Hikata M, Shiraki C, Uematsu I (1982) Polym J 14:205–213
Cheng SZD, Lee SK, Barley JS, Hsu SLC, Harris FW (1991) Macromolecules 24:1883–1889
Shukla P, Muthukumar M (1991) J Polym Sci B Polym Phys 29:1373–1387
Tsabba Y, Rein DM, Cohen Y (2002) J Polym Sci B Polym Phys 40:1087–1094
Block H (1983) Poly(y-Benzyl-L-glutamate) and other glutamic acid containing polymers. Gordon and Breach, New York
Flory PJ (1956) Proceedings of the Royal Society of London. Series A. Math Phys Sci 234:73–89
Doty P, Bradbury JH, Holtzer AM (1956) J Am Chem Soc 78:947–954
Prystupa DA, Donald AM (1993) Macromolecules 26:1947–1955
Cohen Y, Dagan A (1995) Macromolecules 28:7638–7644
Russo PS, Magestro P, Miller WG (1987) In: Russo PS (ed) Reversible polymeric gels and related systems. American Chemical Society, Washington DC, pp 152–180
Miller WG, Kou L, Tohyama K, Voltaggio V (1978) J Polym Sci Polym Symp 65:91–106
Kyu T, Mukherjee P (1988) Liq Cryst 3:631–644
Chowdhury AS, Russo PSJ (1990) Chem Phys 92:5744–5750
Korenaga T, Oikawa H, Nakanishi H (1997) J Macromol Sci Part B: Physics 36:487–501
Sasaki S, Tokuma K, Uematsu I (1983) Polym Bull 10:539–546
Izumi Y, Takezawa H, Kikuta N, Uemura S, Tsutsumi A (1998) Macromolecules 31:430–435
Tadmor R, Dagan A, Cohen Y (1997) Macromol Symp 114:13–22
Chakrabarti S, Miller WG (1984) Biopolymers 23:719–734
Armarego WLF, Perrin DD (1996) Purification of laboratory chemicals, 4th edn. Butterworth-Heinemann, Oxford
Niehoff A, Jurjevic S, Heise A, Menzel H (2009) Macromol Symp 275–276:82–89
Steig S, Cornelius F, Witte P, Staal BBP, Koning CE, Heise A, Menzel H (2005) Chem Commun 43:5420
Teubner M, Strey R (1987) J Chem Phys 87:3195–3200
Schubert KV, Strey R, Kline SR, Kaler EW (1994) J Chem Phys 101:5343–5358
Ilavsky J, Jemian PR (2009) J Appl Crystallogr 42:347–353
Paris O, Li C, Siegel S, Weseloh G, Emmerling F, Riesemeier H, Erko A, Fratzl P (2007) J Appl Crystallogr 40:s466–s470
Hammersley AP, Svensson SO, Hanfland M, Fitch AN, Hausermann D (1996) High Pressure Research 14:235–248
Curtin SA, Deming TJ (1999) J Am Chem Soc 121:7427
Deming TJ (1998) J Am Chem Soc 120:4240
Deming TJ (2000) J Polym Sci Part A: Polym Chem 38:3011
Deming TJ (2006) Adv Polym Sci 202:1–18
Deming TJ (1997) J Am Chem Soc 119:2759
Deming TJ (1997) Adv Mater 9:299–311
Menzel H, Hallensleben ML (1991) Polym Bull 27:89
Kricheldorf HR (1987) Alpha-amino acid-N-carboxyanhydrides and related heterocycles: syntheses, properties, peptide synthesis, polymerization. Springer, Berlin
Habraken GJM, Wilsens KHRM, Koning CE, Heise A (2011) Polym Chem 2:1322–1330
Kuo S-W, Lee H-F, Huang C-F, Huang C-J, Chang F-C (2008) J Polymer Sci, Part A: Polymer Chem 46:3108–3119
Chang YC, Frank CW (1996) Langmuir 12:5824
Elliott A (1967) Poly(R-amino acids): protein models for conformational studies. Marcel Dekker, New York
McKinnon AJ, Tobolsky AV (1968) J Phys Chem 72:1157–1161
Ludwigs S, Krausch G, Reiter G, Losik M, Antonietti M, Schlaad H (2005) Macromolecules 38:7532–7535
Acknowledgments
A. Niehoff thanks the DFG for financial support within the European Graduate School “Microstructural Control in Free-Radical Polymerization” and the DAAD for a 3-month scholarship in the labs of Prof. M. A. Winnik (University of Toronto). Furthermore, financial support by the DFG (project Me 1057/15-3) is acknowledged. A. Mantion thanks the Adolf-Martens e.V. for an “Adolf-Martens Fellowship” and BAM, Berlin, is acknowledged for the financial support. S. Rolf and F. Emmerling are thanked for support during the synchrotron experiments.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 2935 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Niehoff, A., Mantion, A., McAloney, R. et al. Elucidation of the structure of poly(γ-benzyl-l-glutamate) nanofibers and gel networks in a helicogenic solvent. Colloid Polym Sci 291, 1353–1363 (2013). https://doi.org/10.1007/s00396-012-2866-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-012-2866-9