
Abstract
Temperature-induced interchain association and contraction of species in aqueous solutions of charged (MHEC(−)-g-PNIPAAM) and uncharged (MHEC-g-PNIPAAM) modified hydroxyethylcellulose-graft-poly(N-isopropylacrylamide) copolymer have been studied with the aid of turbidimetry and dynamic light scattering (DLS). It was shown that by attaching PNIPAAM chains to the backbone of a hydrophilic cellulose derivative, a strongly temperature-responsive copolymer could be prepared. The results show an intriguing interplay between interchain association and contraction of the multichain species. The transition zone for compression is narrow, and the compaction effect is promoted by a low polymer concentration and charges on the polymer moieties. The findings from DLS revealed two populations of species, namely molecularly dispersed molecules or small clusters and interchain complexes, which exhibit temperature-induced collapse. The magnitude of the cluster contraction can be modulated by changing the polymer concentration and charge density of the copolymer.
Similar content being viewed by others


Introduction
Much attention has been paid to stimuli-sensitive drug delivery systems, which response to external stimuli, such as small changes in pH, ionic strength, and temperature [1–8]. Polymeric micelles and nanobeads belong to this category of drug carriers, and much work has been focused on block copolymers with amphiphilic structure. Hydrophilic blocks can be formed by various materials, including polysaccharides [9–17] and synthetic polymers [18, 19] such as poly(ethylene glycol) [20]. One of the most common studied hydrophobic carriers that is sensitive to temperature is poly(N-isopropylacrylamide) (PNIPAAM) [21]. This polymer exhibits a lower critical solution temperature (LCST) around 32 °C [22] with a heat-induced macroscopic phase separation. This temperature effect is due to the disruption of hydrogen bonding with water and the increasing hydrophobic interactions among isopropyl groups. Temperature-induced coil-to-globule transitions in very dilute aqueous solutions of PNIPAAMs of high molecular weight and narrow molecular weight distributions have been reported [23]. However, even at fairly low temperatures and polymer concentrations, PNIPAAM is known to form interchain associations in aqueous solution [24]. To reduce the tendency of forming interchain complexes and to tune the transition temperature, hydrophilic blocks can be incorporated, or PNIPAAM can be grafted onto the backbone of, e.g., hydrophilic polysaccharide chains. Studies on various polysaccharides, such as carboxymethyl cellulose [9, 12, 25], alginate [13], chitosan [10, 11, 14, 16], and hyaluronic acid [15], with grafted PNIPAAM chains on the backbone have recently been reported.
In the present work, PNIPAAM moieties were grafted onto the hydroxyl groups of the backbone of the uncharged modified hydroxyethylcellulose (MHEC-g-PNIPAAM) sample, or on the anionic hydroxyethylcellulose derivative (sulfo-2-hydroxypropyl substitution) analog for which the abbreviation MHEC(−)-g-PNIPAAM is used. The chemical structures of the polymers are illustrated in Fig. 1. Hydroxyethylcellulose is a biocompatible, biodegradable, and non-toxic hydrophilic polymer with a typical polysaccharide structure [26]. Aqueous solutions of these grafted copolymers are characterized by means of dynamic light scattering and turbidity at different polymer concentrations and temperatures. These experimental methods provide us with information of how temperature, polymer concentration, and charges affect the size of the species and the thermodynamic conditions of the solution.

Schematic illustration of the chemical structures of MHEC, MHEC(−) and PNIPAAM and their graft products. This representation does not imply that all glucosidic units are substituted. The details of the composition of the polymers are given in Table 1
The aim of this investigation is to scrutinize the influence of temperature, polymer concentration, and charges on the intriguing interplay between interchain association and contraction of molecular species at elevated temperatures. It will be shown that intermolecular aggregation is reduced at low polymer concentration and charges on the polymer repress interchain association and favor cluster compression at elevated temperatures. This knowledge is vital in the process of tailor-made particles for drug delivery applications.
Experimental
Materials and preparation of samples
In this study, a hydroxyethylcellulose (HEC) sample with the commercial name Natrosol 250 GR (lot. no. A-0382) acquired from Hercules, Aqualon Division, was used as the hydrophilic polymer and served as the precursor for the synthesis of the copolymers (MHEC-g-PNIPAAM and MHEC(−)-g-PNIPAAM) with grafted PNIPAAM. The degree of substitution of hydroxyethyl groups per repeating anhydroglucose unit for this HEC sample is 2.5 (given by the manufacturer), and the number-average and weight-average molecular masses were determined to be M n = 57 000 g/mol and M w = 400 000 g/mol, respectively, by asymmetric flow field-flow fractionation [27]. The polymer was first desiccated to remove absorbed moisture and then dried under vacuum. The other chemicals are 2-chloroethanol, cerium (IV) ammonium nitrate (CAN), nitric acid solution (1 M; all were purchased from Fluka), and 3-chloro-2-hydroxy-1-propanesulfonic acid sodium salt (from Aldrich) and all chemicals were used as received. N-isopropylacrylamide (NIPAAM, Acros Organics) was recrystallized from a toluene/hexane mixture solvent and dried at room temperature in vacuo prior to use. Regenerated cellulose membrane Spectra/Por 6 with a molecular weight cutoff of 10,000 was purchased from Spectrum Laboratories and utilized in the dialysis.
Synthesis of MHEC-g-PNIPAAM and MHEC(−)-g-PNIPAAM
Modification of HEC
To acquire the grafted copolymer MHEC-g-PNIPAAM with good water-solubility, hydroxyethylation of HEC was carried out to enhance the ethoxyl contents in HEC. A modified sample of hydroxyethylcellulose (MHEC) was prepared by the reaction of HEC with 2-chloroethanol in the presence of a NaOH solution [28]. MHEC was used as the precursor for the succeeding synthesis of the uncharged and negatively charged MHEC-g-PNIPAAM derivatives and because of the modification reaction the values of the molar substitution of the hydroxyethyl groups is increased to 4 (determined by elemental analysis; Atlantic Microlab, Inc).
Synthesis of MHEC-graft-PNIPAAM
Graft polymerization of NIPAAM onto MHEC was performed by using CAN as an initiator under an argon atmosphere at 30 °C following the method published by Wan et al. [28] with some modifications. Redox reaction systems of Ce (IV) with reducing agents (e.g., alcohols, aldehydes, ketones, acids, amines, thiols, etc.) in aqueous solution are well-known initiators for vinyl polymerization of homopolymers and block copolymers; graft copolymers can be easily prepared by this system [29–32].
A certain amount of MHEC was dissolved into pre-degassed distilled water with gently magnetic stirring and argon bubbling for 1 h. To this solution, a degassed cerium (IV) ammonium nitrate (2 × 10-3 M) in nitrite acid (1 M) solution was added and stirred for another 10 min; then N-isopropylacrylamide monomer in degassed water was added, and the polymerization was initiated. The polymerization was conducted at 30 °C for 10 h under an atmosphere of argon. After the polymerization reaction was completed, the solution was diluted with cold water and neutralized with an aqueous solution of 1 M NaOH. To remove unreacted monomer and cerium ions, the resulting mixture was dialyzed repeatedly with fresh distilled deionized water for 2 weeks by using regenerated cellulose with a molecular weight cutoff of about 104 (Spectrum Medical Industries) as the dialyzing membrane. The white solid collected by freeze-drying was further extracted with cold methanol for 24 h to remove rests of the PNIPAAM homopolymer. The MHEC-g-PNIPAAM product was finally dried under vacuum and kept at 4 °C.
Synthesis of MHEC(−)-g-PNIPAAM
MHEC was first mixed with isopropyl alcohol and a 48 % aqueous solution of sodium hydroxide to prepare the slurry. Thereafter, sodium 3-chloro-2-hydroxypropanesulfonate in a 48 % aqueous solution of sodium hydroxide was added, which results in the negatively charged MHEC(−) [33, 34]. The degree of substitution of the charged groups is ca 6.0 mol% analyzed by 1H-NMR. MHEC(−) was then grafted with NIPAAM employing the same procedure as for the uncharged polymer described above.
Characterization of MHEC-g-PNIPAAM and MHEC(−)-g-PNIPAAM
The chemical structure of the graft copolymers were ascertained by their 1H NMR spectra with a Bruker AVANCE DPX 300 NMR spectrometer (Bruker Biospin, Fällanden, Switzerland), operating at 300.13 MHz at 25.0 °C by using heavy water (D2O) as the solvent. The 1H chemical shift in D2O is referred to the residual HDO proton (δ = 4.70 ppm) in D2O. MHEC and HEC show quite similar spectra, but the ratio of the signals of the methylene proton at 3.3–3.4 ppm of MHEC is apparently larger than that of HEC due to the hydroxyethylation of HEC, and it increases the ethoxyl contents in the HEC chain (data is not shown here).
In addition to the typical resonance peak of the HEC chain at 3.5–3.7 ppm, new characteristic signals of the poly(N-isopropylacrylamide) at 1.1 ppm (two methyl groups, -CH3), 3.8 ppm (isopropylmethane groups, -CH-), and 1.5 ppm-2.0 ppm (the main chain CH2 and CH group of PNIPAAM) appear in the proton spectra of the graft derivatives, indicating that the NIPAAM monomer was successfully grafted onto the HEC chain (see Fig. 2). The characteristic data of the samples are collected in Table 1.

a 1H NMR Spectra of MHEC and its graft derivatives: b MHEC-g-PNIPAAM and c MHEC(−)-g-PNIPAAM (300 MHz, 25 °C, and D2O as the solvent)
Turbidimetry
The turbidity measurements were conducted on an NK60-CPA cloud point analyzer from Phase Technology, Richmond, BC, Canada. A detailed description of the equipment and determination of turbidities have been given elsewhere [36]. This apparatus utilizes a scanning diffusive technique to characterize phase changes of the sample with high sensitivity and accuracy. The light beam from an AlGaAs light source, operating at 654 nm, was focused on the measuring sample that was applied onto a special designed glass plate that is coated with a thin metallic layer of very high reflectivity (mirror). Directly above the sample, an optical system with a light-scattering detector continuously monitors the scattered intensity signal (S) of the sample as it is subjected to prescribed temperature alterations. The relation between the signal and the turbidity (τ) is given by the following empirical relationship τ (cm-1) = 9.0 × 10-9S3.751 [35]. In these experiments, the heating rate was set to 0.2 °C/min.
Dynamic light scattering
Dynamic light scattering (DLS) experiments were carried out by means of an ALV/CGS-8 F multi-detector version compact goniometer system, with eight fiber-optical detection units, from ALV-GmbH., Langen, Germany. The beam from a Uniphase cylindrical 22 mW HeNe-laser, operating at a wavelength of 632.8 nm with vertically polarized light, was focused on the sample cell (10-mm NMR tubes, Wilmad Glass Co., of highest quality) through a temperature-controlled cylindrical quartz container (with two plane-parallel windows), which is filled with a refractive index matching liquid (cis-decalin). The temperature in the container is controlled to within ±0.01 °C with a heating/cooling circulator. The polymer solutions were filtered in an atmosphere of filtered air through a 5 μm filter (Millipore) directly into precleaned NMR tubes. The DLS experiments were performed with a temperature gradient (0.2 °C/min) as for the turbidity measurements.
In the solutions of these copolymers, the experimentally recorded intensity autocorrelation function g 2(q,t) (where q is wave vector of magnitude q = (4πn/λ) sin(θ/2), λ is the wavelength of the incident light in a vacuum, θ is the scattering angle, and n is the refractive index of the medium) is directly linked to the theoretically amenable first-order electric field autocorrelation g 1(q,t) through the Siegert [36] relationship \( {g^2}\left( {q,t} \right) = 1 + B|{g^1}\left( {q,t} \right){|^2} \), where B (≤1) is an instrumental parameter.
Elaborate analyses of the correlation functions revealed that, depending on the polymer concentration and temperature, the decays can be described by either the sum of a single exponential and a stretched exponential (Eq. 1a) or by a single stretched exponential (Eq. 1b)
with A f + A s = 1. The parameters A f and A s are the amplitudes for the fast and the slow relaxation time, respectively. The variables τ f and τ se are the relaxation times characterizing the fast and the slow process, respectively. The parameter τ se in Eqs. 1a and 1b is an effective relaxation time, and β (0 < β ≤ 1) is a measure of the width of the distributions of relaxation times. The mean relaxation times are given by
The first term on the right-hand side of Eq. 1a depicts the relaxation of single molecules or small clusters of molecules, whereas the second term represents the relaxation of large association complexes. For both the uncharged and charged grafted copolymers, the decays of the correlation functions could for all the considered polymer concentrations be portrayed by Eq. 1b at temperatures above the LCST. This is also the situation for the uncharged copolymer at temperatures below the LCST when the polymer concentration c ≥ 0.3 wt.%. At this stage, the fraction of large species dominates and the population of molecularly dispersed units is small. At concentrations lower than 0.3 wt.% for the uncharged copolymer, and at all the studied concentrations for the charged copolymer, the correlation functions can be analyzed with the aid of Eq. 1a at temperatures below the LCST. Under these conditions, the growth of interchain complexes is less dominating.
The correlation functions were analyzed with a nonlinear fitting algorithm to obtain best-fit values of the parameters τ se and β appearing on the right-hand side of Eq. 1b or the variables A f, τ f, τ se, and β in Eq. 1a. A fit was considered satisfactory if there were no systematic deviations in the plot of the residuals of the fitted curve.
The fast relaxation mode (τ f) in Eq. 1a is always diffusive (q 2-dependent), whereas the q-dependences of the inverse relaxation time τ -1s are somewhat stronger but less than a q 3-dependence. In view of this, we have as a first approximation chosen to consider all relaxation modes to be diffusive to be able via the Stokes–Einstein relationship to calculate an apparent hydrodynamic radius R h and thereby estimate the sizes of the species
where k B is the Boltzmann constant, T is the absolute temperature, η is the viscosity of the medium, and the mutual diffusion coefficient D = 1/(τ f q 2) for the fast mode or D = 1/(τ s q 2) for the slow mode. The measurements were routinely carried out at eight scattering angles simultaneously, with an accumulation time of 1 min, and the q-dependences of the relaxation times were checked continuously. The abbreviations R h,f and R h,s employed below designate the apparent hydrodynamic radii calculated from the fast and slow relaxation times, respectively.
Results and discussion
In Fig. 3, normalized time correlation function data at a scattering angle of 90o for a high and a low concentration of MHEC-g-PNIPAAM and MHEC(−)-g-PNIPAAM at various temperatures are depicted in form of semilogarithmic plots. In these representations, trivial temperature and solvent viscosity effects on the decays of the correlation functions have been eliminated by introducing the reduced quantity tT/η 0 (η 0 is the solvent viscosity) on the X-axis. A comparison of the decays of the correlation functions clearly unveils a faster relaxation with increasing temperature; this trend is stronger for the samples with the highest concentration and it suggests a temperature-induced compaction of the species.

First-order electric field correlation function versus the reduced variable tT/η 0 for aqueous solutions of MHEC-g-PNIPAAM and MHEC(−)-g-PNIPAAM at two different concentrations and the temperatures indicated
Figure 4 shows temperature and concentration dependences of the apparent hydrodynamic radii for the grafted uncharged copolymer and the charged counterpart at some different polymer concentrations. Let us first discuss the temperature behaviors of the large clusters for all concentrations, except the highest one. At low temperatures, R h,s increases as the polymer concentration is raised (Fig. 4b), indicating that larger aggregates are formed at higher concentrations. However, except for the highest polymer concentration, there is no significant effect of the charged groups on R h,s at low temperatures. The R h,s-temperature curve reveals a distinct contraction transition around 34 °C for the considered concentrations (Fig. 4a). In this temperature region, the grafted PNIPAAM groups become more hydrophobic, causing the chains to self-associate. Therefore, in this temperature-induced transition, there is an omnipresent competition between the growth of aggregates and compaction of the clusters. When the clusters are decorated with charges, it is expected that the tendency of forming aggregates should be reduced because of electrostatic repulsive forces; hence, contraction of the clusters dominates in the transition region, while, for the neutral grafted copolymer, the more pronounced growth of the clusters reduces the magnitude of the contraction effect. This causes the amplitude of the transition to be more marked for the charged copolymer. At temperatures considerably higher than the transition temperature, the apparent hydrodynamic radii decrease slightly for the uncharged copolymer, whereas R h,s for the charged analog increases as the temperature is raised. The reason for this behavior may be that for the uncharged clusters most of the PNIPAAM groups that were initially on the surface of the clusters have already been used to form larger aggregates, causing the neutral polymer to have most of its PNIPAAM groups located inside the clusters at this stage. As a result of this higher density of PNIPAAM segments in the interior of the cluster, contraction of the aggregates dominates for the neutral polymer at the considered high temperatures. For the charged polymer, the repulsive charges initially prevent the clusters from growing, but at high temperatures the PNIPAAM groups become sufficiently hydrophobic to overcome the electrostatic repulsive forces, and larger aggregates start to form. Due to experimental difficulties, cooling curves have not been measured. However, after cooling the system back to low temperatures, the samples revert to their initial state after a period of time. The inset in Fig. 4a illustrates the temperature dependence of the apparent hydrodynamic radii for single molecules or small clusters. Up to the LCST, Rh,f is practically independent of temperature and polymer concentration. These features and the small values of Rh,f suggest that the size of single molecules is probed. The charges cause the polymer coils to expand, causing the sizes of the charged polymer to be slightly larger than what is observed for the neutral polymer. At temperatures close to the LCST, Rh,f increases indicating that some interchain aggregates are formed. At higher temperatures, the slow mode completely dominates the correlation functions, and the fast mode can no longer be detected because the population of small molecules is low. In a previous study [28] on solutions of a graft copolymer, HEC-g-PNIPAAM&PAA, which is composed of HEC as the backbone and a pair of sensitive polymers polyacrylic acid and PNIPAAM as the two grafts, it was shown that the grafted copolymer can self-assemble into micelles with PNIPAAM grafts as the core and HEC/PAA as the corona when the temperature is elevated above the lower critical solution temperature of PNIPAAM.

a Effects of temperature on the apparent hydrodynamic radii for the large clusters for aqueous solutions of MHEC-g-PNIPAAM and MHEC(−)-g-PNIPAAM at the polymer concentrations indicated. The inset plot shows the temperature dependence of the apparent hydrodynamic radii of molecularly dispersed entities or small clusters. b Effects of concentration on the apparent hydrodynamic radius for the large clusters for aqueous solutions of MHEC-g-PNIPAAM and MHEC(−)-g-PNIPAAM at the indicated temperatures
In the case of the highest copolymer concentration (0.5 wt.%), a more intricate picture emerges for the uncharged sample (Fig. 4a). At low temperatures, R h,s of the large interchain complexes decreases slightly with increasing temperature, and at temperatures around the LCST, a drop of the apparent hydrodynamic radius is observed, followed by a plateau at intermediate temperatures and a new drop at still higher temperatures before R hs levels off. This obscure behavior can probably be traced to the intricate interplay between growth of aggregates and the temperature-induced contraction of the multichain associations. The temperature dependence of R h,s of the corresponding charged sample (0.5 wt.%) does not exhibit this complex appearance, but similar features as the other samples. This is ascribed to its less tendency of forming large aggregates at elevated temperatures because of repulsive electrostatic forces.
The ratio between the amplitudes of the slow and the fast mode is displayed in Fig 5. The data in Fig. 5 are taken at a scattering angle of 90o. Generally, the slow mode becomes more dominant at lower scattering angles, and for this system, it dominates completely at very low angles. For both polymers, the slow mode becomes more dominant when the polymer concentration is increased due to the enhanced tendency to form large aggregates at high concentrations. The slow mode is also much more dominant for the neutral polymer than for the charged polymer, since aggregation is reduced in the charged polymer due to the repulsive electrostatic forces. Accordingly, at high concentrations of the neutral polymer, the slow mode completely dominates the correlation functions, and only one mode is observed. Interestingly, for high polymer concentrations of the charged polymer, the slow mode becomes successively less dominant as the temperature is increased. This can be correlated with the contraction of the aggregates (Fig. 4a), as the smaller sizes render the slow mode less dominant. Close to the transition zone, the fast mode becomes more dominant for all systems. This is caused both by a contraction of the large clusters, rendering the slow mode to be less dominant, and by an incipient aggregation of the single molecules of the fast mode into small aggregates (see inset in Fig. 4a). After the transition zone, very few single molecules are left in the samples causing the large and compact clusters to completely dominate the correlation functions, and only one relaxation mode is observed.

The effect temperature of the ratio between the amplitudes of the fast and slow mode at the indicated polymer concentrations at a scattering angle of 90o for a MHEC-g-PNIPAAM and b MHEC(−)-g-PNIPAAM
Figure 6a illustrates the temperature dependence of the turbidity for different concentrations of the uncharged and charged copolymers. The curves reveal a sharp transition around 32 °C, followed by a plateau-like region at higher temperatures. The transition becomes more accentuated at higher polymer concentration, and the plateau-like regions at elevated temperatures assume higher values of the turbidity for the concentrations of the uncharged copolymer than for the corresponding concentrations of the charged copolymer. The turbidity of a sample is affected by the number of particles, the size of the particles, and the compactness of the particles (i.e., the ratio between the refractive index of the particle (n p ) and the refractive index of the solvent (n 0)). Therefore, both a contraction of the particles and enhanced aggregation can cause an increase in the turbidity. To facilitate a direct comparison of the features of the uncharged and charged copolymer, the DLS and turbidity data for a single polymer concentration (0.1 wt.%) are displayed in Fig. 7. For both polymers, the contraction of the clusters at about 34 °C causes a corresponding increase in the turbidity values. At temperatures above the transition region, the turbidity of the neutral polymer continues to increase while for the charged polymer it flattens out at elevated temperatures. At the same time, R h,s is decreasing for the neutral polymer and increasing for the charged polymer. This indicates that the shrinking of the aggregates causes them to become much more compact causing the higher turbidity values.

a Illustration of the temperature effect on the turbidity behavior for the uncharged (MHEC-g-PNIPAAM) and charged (MHEC(−)-g-PNIPAAM) copolymer at different concentrations. b Effects of polymer concentration on the cloud point, which is determined from the onset of the increase of the turbidity while raising the temperature

Temperature dependences of the apparent hydrodynamic radii and the turbidity for 0.1 wt.% solutions of MHEC-g-PNIPAAM and MHEC(−)-g-PNIPAAM
The turbidity values are overall lower for the charged copolymer than for the neutral polymer. At low temperatures, the repulsive electrostatic forces prevent the clusters of the charged polymer from becoming as compact as the clusters of the neutral polymer. This effect might also contribute to the lower turbidity values of the charged polymer at elevated temperatures, but, in this region, the greater size of the clusters formed by the neutral polymer will also contribute to the difference in turbidity.
To obtain further information about the thermodynamic conditions of the systems, the cloud point was determined. The temperature at which the first deviation of the scattered intensity from the baseline occurred was taken as the cloud point (CP) of the corresponding solution. The effect of polymer concentration on CP is depicted in Fig. 6b for the two copolymers. The values of CP are higher for the charged copolymer and a weak concentration dependence of CP is found. The effect of polymer concentration on CP is much stronger for the uncharged counterpart. These differences in CP behavior of the copolymers are again a manifestation of the impact of the electrostatic interactions to weaken the interchain association.
To see the effect of the screening of the electrostatic interactions, salt (NaCl) was added to the system. The effect of salt addition on the turbidity of the two polymers is displayed in Fig. 8. The same transition in the turbidity around LCST is observed when salt is added to the system, but the turbidity values are generally much higher in the presence of salt. The effect of salt addition is especially strong for the charged polymer at elevated temperatures, where is assumes much higher values than what is observed in the absence of salt. Interestingly, the turbidity values are even higher than what is observed for the neutral polymer either in the absence or presence of salt. The reason for this is discussed below.

The effect of salt addition on the temperature dependence of the turbidity at a polymer concentration of 0.1 wt.% of MHEC-g-PNIPAAM and MHEC(−)-g-PNIPAAM
The effect of salt on R h for the two polymers is displayed in Fig. 9. As for the turbidity values (Fig. 8), the differences between the samples containing 0.05 and 0.1 M NaCl is small, indicating that most of the charges has been screened out already at 0.05 M NaCl. For both polymers, salt addition induces only minor changes in the values of R h,f. The neutral polymer exhibits an enhanced tendency to form large aggregates in the presence of salt (Fig. 9a). Salt induces poorer thermodynamic conditions for the polymer (salting-out effect), which lead to the formation of larger polymer aggregates. The charged polymer is not significantly affected by the addition of salt at low temperatures (Fig. 9b). The sizes of the large aggregates (R h,s) are close to what is observed in the absence of salt for both the neutral and the charged polymers in this temperature region, but lower than what is observed for the neutral polymer in the presence of NaCl. This indicates that the charged polymer is less prone to salting-out effects than the neutral polymer. The reason for this might be that in the charged polymer the salt ions are more likely to be located close to the charged groups, reducing the salting-out effect of the remaining polymer chain. R h,s of the charged polymer in the presence of salt exhibits a transition region, which occurs at a slightly lower temperature than in the absence of salt. However, in the presence of salt, the clusters become larger instead of smaller in the transition region. As discussed above, there is a competition between shrinking and aggregation of the large clusters. For the neutral polymer, both in the absence and presence of salt, and for the charged polymer in the absence of salt, shrinking dominates in the transition region. When the charges are screened, the charged polymer should in principle behave in a similar manner as the neutral polymer. The growth of the aggregates during the transition zone could either be caused by enhanced salting-out effects at higher temperatures, which should give aggregate sizes closer to what is observed for the neutral polymer, or enhanced aggregation compared with the neutral polymer because the charged polymer contain more PNIPAAM groups than the neutral polymer (see Table 1). The very high turbidity values observed at high temperatures for the charged polymer in the presence of salt (Fig. 8) indicate that at these conditions the clusters are much more compact than for the neutral polymer. It is therefore likely that when the charges are screened out, the higher number of PNIPAAM groups causes the charged polymer to form larger and more compact clusters than the neural one. After the transition region, the sizes of these clusters start to shrink in a similar way as is observed for the neutral polymer.

The effect of salt addition on the temperature dependence of R r,f and R h,s at a polymer concentration of 0.1 wt.% of MHEC-g-PNIPAAM and MHEC(−)-g-PNIPAAM
Conclusions
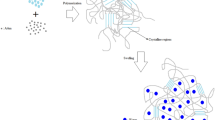
In this work, PNIPAAM chains have been grafted onto the backbone of HEC with (MHEC(−)-g-PNIPAAM) and without (MHEC-g-PNIPAAM) charges. The properties of the hydrophilic HEC are drastically altered when PNIPAAM sequences are attached to the polymer. The DLS measurements disclose a kind of temperature-induced coil-to-globule transition of the interchain structures, with a more pronounced compression for the charged copolymer. The picture that emerges from the DLS and turbidity results is that during the temperature raise there is an omnipresent competition between the growth of aggregates because of enhanced hydrophobic interactions and contraction of the species due to the packing of the hydrophobic segments to avoid exposure to water. The results clearly show that the temperature-induced interplay between interchain association with cluster growth and compression of the complexes is governed by polymer concentration and electrostatic interactions. Although the charges incorporated on the cellulose derivative reduce the tendency of the copolymer to form large clusters, especially at high polymer concentrations, the charges do not protect the molecules from forming interchain associations. This indicates that there are PNIPAAM chains sticking out from the HEC backbone, which facilitate interchain association in spite of the charges attached to the polysaccharide chains. However, the results at low polymer concentration have demonstrated that strong compression occurs for copolymer chains decorated with charges. A schematic illustration of the effects of temperature on the charged and the neutral polymers are displayed in Fig. 10. This demonstrates that the delicate interplay between aggregation and compression of the species can be tuned by altering polymer concentration, temperature, and charge density. This type of temperature-sensitive beads can be a potential candidate for drug delivery applications.

Schematic illustration of effect of temperature on the aggregation and contraction behavior in solutions of MHEC-g-PNIPAAM and MHEC(−)-g-PNIPAAM
References
Pelton R (2000) Adv Colloid Interface Sci 85:1
Jones CD, Lyon LA (2000) Macromolecules 33:8301
Peppas NA, Bures P, Leobandung W, Ichikawa W (2000) Eur J Pharm Biopharm 50:27
Zrinyi M (2000) Colloid Polym Sci 16:2623
Ju HK, Kim SY, Lee YM (2001) Polymer 42:6851
Nayak S, Lyon LA (2004) Chem Mater 16:2623
Nayak S, Lee H, Chmielewski LA, Lyon LA (2004) J Am Chem Soc 126:10258
Kjøniksen A-L, Zhu K, Pamies R, Nyström B (2008) J Phys Chem B 112:3294
Vasile C, Marinescu C, Vornicu R, Staikos G (2003) J Appl Polym Sci 87:1383
Lee SB, Dong IH, Cho SK, Kim SJ, Lee YM (2004) J Appl Polym Sci 92:2612
Cho JH, Kim S-H, Park KD, Jung MC, Yang WI, Han SW, Noh JY, Lee JW (2004) Biomaterials 25:5743
Vasile C, Bumbu GG, Dumitriu RP, Staikos G (2004) Eur Polym J 40:1209
Kim MH, Kim J-C, Lee HY, Kim JD, Yang JH (2005) Colloids Surf B 46:57
Chen J-P, Cheng T-H (2006) Macromol Biosci 6:1026
Ha DI, Lee SB, Chong MS, Lee YM, Kim SY, Park YH (2006) Macromol Res 14:87
Sun G, Zhang X-Z, Chu C-C (2008) J Mater Sci Mater Med 19:2865
Li X, Yin M, Zhang G, Zhang F (2009) Chin J Chem Eng 17:145
Tauer K, Gau D, Schulze S, Vökel A, Dimova R (2009) Colloid Polym Sci 287:299
Liu R, Fraylich M, Saunders BR (2009) Colloid Polym Sci 287:627
Topp MDC, Dijkstra P, Talsma H, Feijen I (1997) Macromolecules 30:8518
Stile RA, Burghardt WR, Healy KE (1990) Macromolecules 32:7370
Schild HK (1992) Prog Polym Sci 17:163
Wu C, Zhou S (1995) Macromolecules 28:8381
Pamies R, Zhu K, Kjøniksen A-L, Nyström B (2009) Polym Bull 62:487
Kim JH, Lee SB, Kim SJ, Lee YM (2002) Polymer 43:7549
Nyström B, Kjøniksen A-L, Beheshti N, Zhu K, Knudsen KD (2009) Soft Matter 5:1328
Beheshti N, Zhu K, Kjøniksen AL, Nyström B (2008) Colloid Surf A Physicochem Eng Asp 328:79
Wan S, Jiang M, Zhang G (2007) Macromolecules 40:5552
Misra GS, Bajpai VDN (1982) Prog Polym Sci 8:61
Sarac AS (1999) Prog Polym Sci 24:1149
Motokawa R, Morishita K, Koizumi S, Nakahira T, Annaka M (2005) Macromolecules 38:5748
Derkaoui SM, Avramoglou T, Barbaud C, Letourneur D (2008) Biomacromolecules 9:3033
Miyajima T, Kitsuki T, Kita K, Kamitani H, Yamaki K (1999) US Patent 5,891,450
Antonio MR (1998) Polymer 39:3115
Kjøniksen A-L, Laukkanen A, Galant C, Knudsen KD, Tenhu H, Nyström B (2005) Macromolecules 38:948
Siegert AJF Radiation Laboratory Report No. 465, Massachusetts Institute of Technology, Cambridge, MA, 1943
Acknowledgment
This work was financially supported from the Research Council of Norway within the FRINAT program, project no. 177556/V30.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Phan, H.T.T., Zhu, K., Kjøniksen, AL. et al. Temperature-responsive self-assembly of charged and uncharged hydroxyethylcellulose-graft-poly(N-isopropylacrylamide) copolymer in aqueous solution. Colloid Polym Sci 289, 993–1003 (2011). https://doi.org/10.1007/s00396-011-2423-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-011-2423-y